
Samenvatting
- Erfelijke stofwisselingsziekten bestaan uit een divers palet van ruim 800 zeldzame aandoeningen.
- Stofwisselingsziekten waren voorheen vooral het domein van de kindergeneeskunde vanwege het erfelijke karakter en de vaak beperkte levensverwachting.
- Niet alle stofwisselingsziekten openbaren zich tijdens de kinderleeftijd; een significant deel manifesteert zich voor het eerst op volwassen leeftijd.
- Het aantal volwassen patiënten met een erfelijke stofwisselingsziekte neemt sterk toe door verbeterde zorg en uitbreiding van de diagnostische mogelijkheden. Deze toename zal ertoe leiden dat meer specialismen met deze groep patiënten geconfronteerd zullen gaan worden.
- De volwassen zorg kent haar eigen specifieke uitdagingen, zoals de zorg rond de zwangerschap van een patiënt met een stofwisselingsziekte.
- Langdurig vasten, bijvoorbeeld rond een operatie, kan leiden tot een levensbedreigende metabole ontregeling als er geen juiste voorzorgsmaatregelen worden getroffen.
- De groep Internisten voor Volwassenen met een Erfelijke Stofwisselingsziekte (INVEST) is een initiatief van internisten ter bevordering van de zorg voor volwassen patiënten met een stofwisselingziekte.
Leerdoelen
- Erfelijke stofwisselingsziekten omvatten een diverse groep aandoeningen van het metabolisme.
- De zorg voor patiënten met een erfelijke stofwisselingsziekte was voorheen voorbehouden aan de kinderarts.
- Het aantal volwassen patiënten met een stofwisselingsziekte neemt sterk toe.
- De groei van het aantal volwassen patiënten zal leiden tot nieuwe uitdagingen die zich niet zullen beperken tot 1 medische discipline.
- De groep Internisten voor Volwassenen met een Erfelijke Stofwisselingsziekte (INVEST) ziet het als haar taak de zorg voor volwassen patiënten met een stofwisselingsziekten te verbeteren en op te treden als vraagbaak voor andere specialismen.
artikel
Erfelijke stofwisselingsziekten zijn een groep aangeboren aandoeningen waarbij er sprake is van een defect in een enzym, cofactor of transporter. Hierdoor ontstaat er een stoornis in het koolhydraat-, vet- of eiwitmetabolisme, of in de synthese of afbraak van complexe moleculen. Over deze aandoeningen is eerder in het NTvG geschreven.1-5 Door dit erfelijke defect ontstaat ophoping van substraat of een tekort aan een ander molecuul, bijvoorbeeld aan het eindproduct van een enzymatische reactie. Het substraat kan toxisch zijn en zo schade toebrengen, bijvoorbeeld cerebrale schade door hoge spiegels van fenylalanine bij fenylketonurie (PKU). Bij sommige aandoeningen kan continue stapeling van substraat leiden tot orgaanvergroting, bijvoorbeeld hepatosplenomegalie bij de ziekte van Gaucher.
Er zijn momenteel ruim 800 erfelijke metabole ziekten beschreven, waarvan de meeste zeldzaam tot uiterst zeldzaam zijn. Er wordt echter geschat dat ten minste 1 op de 2500 levendgeborenen een stofwisselingsziekte heeft.6,7 Dat betekent dat er ongeveer 6700 mensen in Nederland aangedaan zijn, wat overeenkomt met 1 patiënt per huisarts. Aangezien deze getallen gebaseerd zijn op diagnoses die op de kinderleeftijd gesteld zijn – en niet alle stofwisselingsziekten zich voor het eerst op deze leeftijd manifesteren – is de verwachting dat deze prevalentiecijfers in werkelijkheid nog hoger zijn.
Voorheen waren erfelijke stofwisselingsziekten bij uitstek het terrein van de kinderarts en dit specialisme erkent dan ook het aandachtsgebied metabole ziekten. Door verbeterde zorg, uitbreiding van de diagnostische mogelijkheden en toegenomen bewustwording ('awareness') van erfelijke metabole aandoeningen neemt het aantal volwassenen met een erfelijke stofwisselingsziekte sterk toe. Het zeldzame voorkomen en de specifieke problemen die zich op volwassen leeftijd voordoen maakt dat gespecialiseerde zorg voor hen belangrijk is.
In dit artikel bespreken wij het spectrum van erfelijke stofwisselingsziekten op volwassen leeftijd in Nederland en de aandachtspunten bij de zorg van patiënten met deze veelal complexe aandoeningen.
Omvang van groep volwassenen
De zorg voor volwassenen met een erfelijke stofwisselingsziekte is geconcentreerd in een aantal academische centra. Deze zorg neemt toe in omvang. Een steekproef in 3 van deze centra (AMC, Erasmus MC en Radboudumc) laat zien dat er in 2009 gezamenlijk 846 volwassen patiënten bekend waren op de poliklinieken Erfelijke Stofwisselingsziekten. In 2012 is deze populatie – die het complete palet aan erfelijke stofwisselingsziekten omvat (figuur 1) – met 48% gegroeid. Deze patiëntenpopulatie is relatief jong (gemiddelde: 38 jaar; spreiding: 16-69) en de diagnose is bij een substantieel deel pas op volwassen leeftijd gesteld. Dit geldt voor 95% van de patiënten met lysosomale stapelingsziekten en voor 20% van de patiënten met andere stofwisselingsziekten (gegevens van het AMC-cohort).
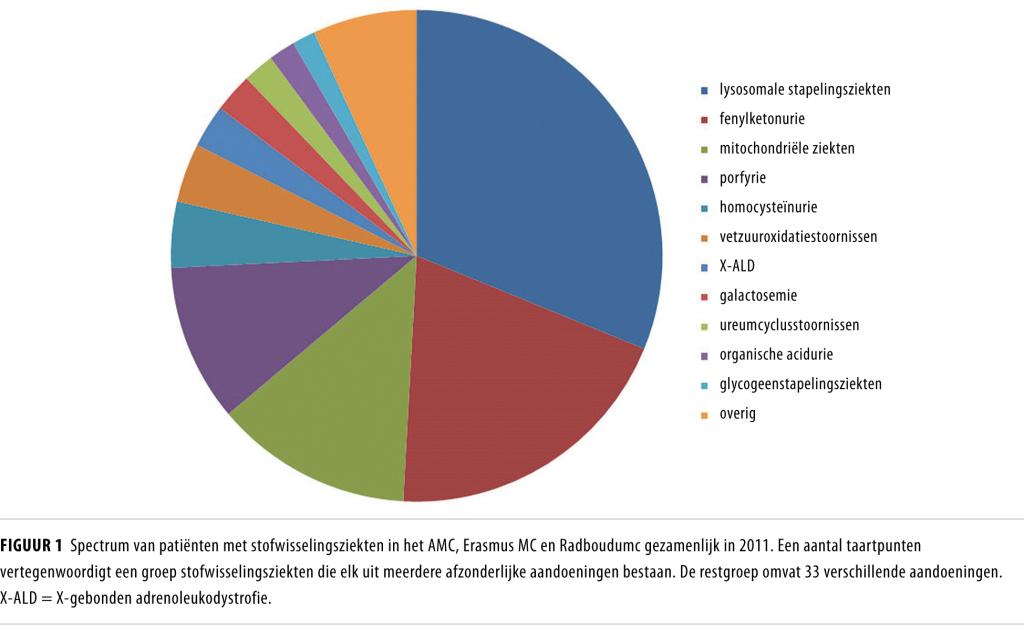
Momenteel is de bekendste stofwisselingsziekte PKU, ook omdat dit in Nederland de eerste aandoening was waarop werd gescreend bij pasgeborenen, vanaf 1974. Momenteel wordt bij de hielprikscreening gescreend op 20 aandoeningen, waaronder 14 erfelijke stofwisselingsziekten (zie voor een overzicht Visser et al.2). Sinds de uitbreiding van de hielprik in 2007 worden jaarlijks 60-100 pasgeborenen gediagnosticeerd met een erfelijke stofwisselingsziekte.8 Onderzoek heeft aangetoond dat een deel van deze patiënten niet ontdekt zou zijn wanneer de diagnostiek had plaatsgevonden op louter klinische gronden.9 De toename van het aantal geïdentificeerde kinderen met een stofwisselingsziekte zal vanaf 2024 dus ook leiden tot transitie van deze patiënten naar de zorg voor volwassenen.
Zorg voor groep volwassenen
Zoals gezegd vindt de eerste manifestatie van een erfelijke stofwisselingsziekte niet noodzakelijkerwijs plaats op de neonatale leeftijd of kinderleeftijd. De ziekte van Fabry, een lysosomale stapelingsziekte, openbaart zich bijvoorbeeld meestal tussen de leeftijd van 10 en 30 jaar.10 Maar ook aandoeningen die tot acute ontregelingen kunnen leiden, zoals ureumcyclusdefecten of vetzuuroxidatiestoornissen, manifesteren zich soms pas voor het eerst op de volwassen leeftijd.
Wanneer moet er nu gedacht worden aan een erfelijke stofwisselingsziekte? Vanwege de lage a-priorikans dat iemand in de algemene bevolking een stofwisselingsziekte heeft, is het verstandig op zoek te gaan naar het zogenaamde 'plus-teken'. Zo is de a-priorikans dat een patiënt met diabetes mellitus een mitochondriële aandoening heeft erg klein. Wanneer er nu echter sprake is van diabetes mellitus plusdoofheid, is de kans op een mitochondriële ziekte aanzienlijk groter, bijvoorbeeld op 'maternally inherited diabetes and deafness' (MIDD).11 Een overzicht van mogelijke presentaties van erfelijke stofwisselingsziekten op volwassen leeftijd (metplus-teken) is gegeven in tabel 1; deze tabel dient als voorbeeld en is zeker niet volledig.

Toevalsbevindingen
Het gevaar van het routinematig screenen op stofwisselingsziekten zonder aanwezigheid van het plus-teken levert het gevaar van fout-positieve bevindingen op. Bovendien geldt voor veel aandoeningen dat de ernst van de ziekte varieert. Sommige individuen hebben wel een genetische afwijking, maar niet noodzakelijkerwijs de ziekte. Dit is bijvoorbeeld bij de ziekte van Fabry het geval. Een ander voorbeeld betreft de neonatale screening: hoewel dit tot relatief weinig fout-positieve bevindingen leidt,8 komt het fascinerend genoeg ook wel eens voor dat de moeder een stofwisselingsziekte blijkt te hebben die wordt vastgesteld via het bloed van de pasgeborene. De pasgeborene is dan gezond. Voorbeelden hiervan zijn methylcrotonyl-CoA-carboxylase(3-MCC)-deficiëntie, carnitinedeficiëntie, glutaaracidurie type 1 en partiële biotinidasedeficiëntie.12,13
Het klinisch belang van deze 'metabole incidentalomen' of 'DNA-incidentalomen' is onduidelijk, maar zal door de toename van metabole en genetische screening veel vragen en kosten opleveren.14
Noodopvang
Een aantal stofwisselingsziekten, zoals acute porfyrie, vetzuuroxidatiestoornissen, glycogeenstapelingsziekten, organische acidurieën en ureumcyclusstoornissen, kunnen zorgen voor een acute ontregeling en vervolgens leiden tot een levensbedreigende situatie. Dergelijke ernstige ontregelingen kunnen veroorzaakt worden door een katabole situatie door verminderde voedselinname, bijvoorbeeld bij een 'simpele' intercurrente virale infectie, bevalling of operatie. Hierbij is een patiënt in hoge mate afhankelijk van endogene energieproductie middels glycogenolyse, proteolyse of lipolyse. Juist wanneer er een stoornis is in 1 van deze paden kan een ernstige hypoglykemie, acidose of lactaatacidose, of hyperammoniëmie ontstaan.
Hoewel deze ontregelingen vroeger een zeer hoge mortaliteit kenden, is de behandeling tegenwoordig aanzienlijk verbeterd, waardoor kinderen de volwassenheid kunnen bereiken.15 Dat betekent dat ook andere specialismen in toenemende mate geconfronteerd zullen worden met patiënten met een metabole ontregeling of een dreiging daarop. Dit betreft niet alleen huisartsen en internisten – die de eersten zijn die deze patiënten zullen opvangen tijdens zo'n ontregeling – maar ook de snijdende specialismen. Net als bij een intercurrente infectie leidt vasten rond een operatie tot een katabole status en kan daarmee een metabole ontregeling uitgelokt worden.
In figuur 2 staan de algemene principes van de behandeling van stofwisselingsziekten. In tabel 2 worden vervolgens de behandeling én klinische presentatie van patiënten met de meest voorkomende acute ontregelingen specifiek toegelicht. Uit deze tabel valt af te leiden dat er bij een vermoeden van een acute metabole ontregeling vrijwel altijd direct gestart dient te worden met glucose-infusie. Het onderliggende principe van glucose-infusie is enerzijds substitutietherapie (bij stofwisselingsziekten waar hypoglykemie op de voorgrond staat) en anderzijds substraatreductietherapie door voldoende calorieën aan te bieden ter verdere preventie van katabolisme en daarmee samenhangend het ontstaan van toxische metabolieten. Uiteraard is frequente monitoring met controle van elektrolytenwaarden noodzakelijk om bijvoorbeeld hyponatriëmie te voorkomen.

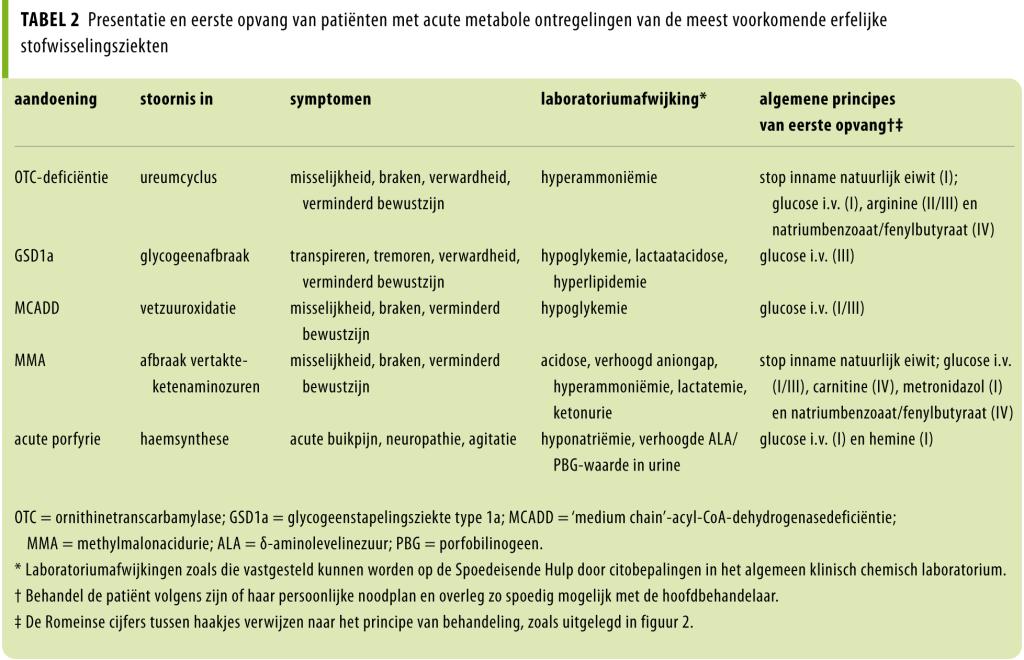
Algemene richtlijnen zijn dan ook een simplificatie. Zo zal voor patiënten met een ureumcyclusdefect of stoornis in de aminozuurafbraak bij ontregeling initieel gestopt worden met de eiwitinname, maar zal vrij snel weer eiwit geïntroduceerd moeten worden om aan de basale behoefte te voldoen. Dit vergt continue monitoring en overleg met een specialist erfelijke stofwisselingsziekten. Patiënten met een erfelijke stofwisselingsziekte die kunnen ontregelen moeten dan ook in het bezit zijn van een noodprotocol met daarin informatie over hoe te handelen bij een al dan niet dreigende metabole ontregeling en met wie in contact te treden. Bij operatieve ingrepen dienen perioperatieve maatregelen te worden getroffen die gericht zijn op preventie van een katabole status.
Zwangerschap
Specifieke zorg is tevens noodzakelijk rond de zwangerschap. Dankzij verbeterde overleving hebben patiënten met een stofwisselingsziekte ook de mogelijkheid om zelf zwanger te worden. Dit vereist vaak genetische preconceptionele counseling door een klinisch geneticus en begeleiding door een gespecialiseerde internist, diëtist en gynaecoloog.
Voor veel erfelijke aandoeningen is de wetenschappelijke bewijsvoering of een zwangerschap wel veilig is voor de ongeboren vrucht of voor de moeder met een stofwisselingsziekte slechts gebaseerd op patiëntbeschrijvingen.1,16 Deze laten bijvoorbeeld zien dat metabole ontregelingen bij de moeder voornamelijk optreden in het kraambed wanneer de proteolyse maximaal is door involutie van de uterus. Een adequate koolhydraatinname in combinatie met eventuele dieetaanpassingen en nauwgezette observatie van symptomen en plasma- of urinemetabolieten is daarom van essentieel belang, niet alleen tijdens de partus maar ook in het kraambed.16
Voor patiënten met PKU is er wel onomstotelijk bewijs dat voorafgaand aan en tijdens de zwangerschap een strikt fenylalaninebeperkt dieet en nauwgezette controle van de fenylalaninewaarden noodzakelijk zijn om ernstige congenitale afwijkingen bij de foetus te voorkomen. Deze afwijkingen, ook wel maternale PKU genoemd, worden gekenmerkt door microcefalie, hartklepgebreken en een onherstelbare verstandelijke beperking.17 Episodes van verminderde voedselinname, zoals bij hyperemesis gravidarum, kunnen gepaard gaan met toegenomen eiwitafbraak (katabolisme) en daardoor met toxische stijgingen van de fenylalanineconcentratie. Dit dient beschouwd te worden als een noodsituatie. Directe klinische behandeling met anti-emetica, adequate voeding of sondevoeding, of intraveneus glucose is noodzakelijk om katabolisme te voorkomen.
Nieuwe behandelingen
Hoewel de diversiteit van verschillende stofwisselingsziektes enorm is, zijn de algemene principes van behandeling betrekkelijk uniform voor veel aandoeningen (zie figuur 2). Zo bestaat de behandeling van patiënten met PKU uit een fenylalaninebeperkt dieet ter reductie van dit toxisch substraat. Deze behandeling is tegenwoordig veilig, omdat we beschikken over aminozuurpreparaten zonder fenylalanine die een dreigend tekort aan essentiële aminozuren, vitaminen en mineralen voorkomen.
Een beperkt aantal patiënten profiteert tevens van behandeling met tetrahydrobiopterine (sapropterine), een cofactor van fenylalaninehydroxylase, waardoor de enzymactiviteit verbeterd wordt.18 Vooral van het dieet is aangetoond dat bij goede compliantie de cognitieve ontwikkeling van patiënten met PKU ongestoord verloopt, waardoor ze volwaardig kunnen deelnemen aan de maatschappij, zoals de meeste patiënten met een stofwisselingsziekte. Zoals al eerder beargumenteerd in het NTvG dienen de aminozuursupplementen als medicijn beschouwd te worden; deze supplementen worden, weliswaar in een andere samenstelling, ook gebruikt bij de behandeling van patiënten met organische acidurie of klassieke homocystinurie. Een verkeerde indicatiestelling of verwisseling is potentieel levensgevaarlijk.3
De opkomende behandelingen van patiënten met erfelijke stofwisselingsziekten zijn met name gericht op verbetering van de residuele enzymactiviteit. De eerste successen zijn geboekt bij lysosomale stapelingsziekten. Dit kan met enzymvervangende therapie, zogenaamde 'chaperone'-therapie en gentherapie (zie figuur 2).
Enzymvervangende therapie voor patiënten met de infantiele vorm van zure-maltasedeficiëntie (ziekte van Pompe; een glycogeenstapelingsziekte of lysosomale stapelingsziekte die leidt tot progressieve spierzwakte) heeft bijvoorbeeld geleid tot een substantiële verbetering van de prognose.19 Ook allogene stamcel- of orgaantransplantatie is in feite een vorm van gentherapie, die bij vele aandoeningen is geprobeerd en bij een aantal succesvol is gebleken, zoals levertransplantatie bij patiënten met tyrosinemie of stamceltransplantatie bij onder andere patiënten met metachromatische leukodystrofie.20,21 Meestal betreft het hier ingrepen die op de kinderleeftijd worden verricht, maar soms gebeurt dit ook bij volwassenen.
Organisatie van zorg
Het groeiend aantal volwassen patiënten met een stofwisselingsziekte – door verbeterde diagnostiek en behandelingsmogelijkheden – en de uitdagingen die daaruit voortvloeien vragen om een gecentraliseerde aanpak. Daarom hebben de internisten gespecialiseerd in erfelijke stofwisselingsziekten uit de verschillende academische centra, inclusief Universitair Ziekenhuis Leuven, zich in 2011 verenigd in de groep Internisten voor Volwassenen met een Erfelijke Stofwisselingsziekte (INVEST); deze groep is recent ondergebracht bij de Nederlandse Vereniging voor Endocrinologie.
De INVEST-groep heeft zich verschillende doelen gesteld. Ten eerste dient de zorg voor de groeiende groep volwassen patiënten in Nederland gewaarborgd te worden. Internisten gespecialiseerd in erfelijke stofwisselingsziekten hebben een gezamenlijk transitiespreekuur met de kinderarts-metabole ziekten om de overgang van de adolescente patiënt met een stofwisselingsziekte naar de volwassen polikliniek zo soepel mogelijk te laten verlopen.
Bovendien worden landelijke richtlijnen en noodprotocollen opgesteld voor patiënten met een al dan niet dreigende acute metabole ontregeling. De patiëntenorganisatie Volwassenen, Kinderen en Stofwisselingsziekten (VKS) heeft al in samenwerking met kinderartsen, diëtisten en leden van de INVEST-groep verschillende zorgpaden voor zowel patiënten als professionals opgesteld.
Aangezien patiënten dikwijls naar hun eigen regionale ziekenhuis gaan met een acute ontregeling, is het belangrijk dat ook internisten in niet-academische centra deze noodprotocollen kunnen raadplegen. Patiënten beschikken meestal zelf over zo'n noodprotocol en deze zal in de toekomst ook online beschikbaar worden gesteld (www.investof.nl). Via dezelfde site – die momenteel nog in ontwikkeling is – is nu te zien welke reguliere centra, expertisecentra en specialisten benaderbaar zijn voor overleg.
Daarnaast zal de groep zich toeleggen op scholing, het verbeteren van de bewustwording van het vóórkomen van erfelijke stofwisselingsziekten, en het initiëren van en deelnemen aan nationale en internationale studies.
Conclusie
Erfelijke stofwisselingsziekten bestaan uit een divers palet van verschillende aandoeningen; onze kennis over een groot deel hiervan is nog onontgonnen. Een verbetering van deze kennis zal er in toenemende mate voor zorgen dat stofwisselingsziekten zich niet meer zullen beperken tot het domein van de kinderarts, maar dat ook andere specialismen steeds meer geconfronteerd zullen worden met dit fascinerende vakgebied. De INVEST-groep heeft als doel daarbij als een baken te dienen, enerzijds voor de volwassen patiënt die kan vertrouwen op hoogwaardige zorg en anderzijds voor collega-medici met vragen of problemen rond de zorg voor deze patiënten.
Literatuur
Van Spronsen FJ, Molendijk H, Erwich JJ, Smit GP. Erfelijke stofwisselingsziekten en zwangerschap: consequenties voor moeder en kind. Ned Tijdschr Geneeskd. 2003;147:235-40 Medline.
Visser G, van Spronsen FJ, de Sain-van der Velden MG, Blom HJ, Wijburg FA. Uitgebreide neonatale hielprikscreening op stofwisselingsziekten in Nederland. Evaluatie van de eerste 2 jaar. Ned Tijdschr Geneeskd. 2009;153:B360 Medline.
Van der Wiel AM, Janssen M, Hollack C, Langendonk JG. Complicaties door verwisseling aminozuurpreparaten. Ned Tijdschr Geneeskd. 2013;157:A5183 Medline.
Gubbels NP, Sijbrand MH, Onstenk R. Zwarte urine of zwart oogwit? Denk aan alkaptonurie. Ned Tijdschr Geneeskd. 2014;158:A6684.
Bangma HR, Smit GP, Kuks JB, Grevink RG, Wolffenbuttel BH. Twee patiënten met een mitochondriale myopathie. Ned Tijdschr Geneeskd. 2008;152:2298-301 Medline.
Dionisi-Vici C, Rizzo C, Burlina AB, et al. Inborn errors of metabolism in the Italian pediatric population: a national retrospective survey. J Pediatr. 2002;140:321-7 Medline. doi:10.1067/mpd.2002.122394
Applegarth DA, Toone JR, Lowry RB. Incidence of inborn errors of metabolism in British Columbia, 1969-1996. Pediatrics. 2000;105:e10 Medline. doi:10.1542/peds.105.1.e10
Lanting CI, Rijpstra A, Verkerk PH. Monitor en evaluatie van de nenonatale hielprikscreening bij kinderen geboren in 2011. Rapportnummer: TNO/CH 2013 R10545. Leiden: TNO; 2013.
Wilcken B, Wiley V, Hammond J, Carpenter K. Screening newborns for inborn errors of metabolism by tandem mass spectrometry. N Engl J Med. 2003;348:2304-12 Medline. doi:10.1056/NEJMoa025225
Linthorst GE, Hollak CE, Bosman DK, Heymans HS, Aerts JM. De ziekte van Fabry: op weg naar een behandeling. Ned Tijdschr Geneeskd. 2000;144:2391-5 Medline.
'T Hart LM, Lemkes HH, Heine RJ, Stolk RP, Feskens EJ, Jansen JJ, et al. Prevalence of maternally inherited diabetes and deafness in diabetic populations in The Netherlands. Diabetologia. 1994;37:1169-70. Medline
Lund AM, Hougaard DM, Simonsen H, et al. Biochemical screening of 504,049 newborns in Denmark, the Faroe Islands and Greenland--experience and development of a routine program for expanded newborn screening. Mol Genet Metab. 2012;107:281-93 Medline. doi:10.1016/j.ymgme.2012.06.006
Vilarinho L, Rocha H, Sousa C, et al. Four years of expanded newborn screening in Portugal with tandem mass spectrometry. J Inherit Metab Dis. 2010;33(Suppl 3):S133-8 Medline. doi:10.1007/s10545-010-9048-z
Westerink J, Spiering W. Consequenties van 'next-generation sequencing'. Wie kennis vermeerdert, vermeerdert smart? Ned Tijdschr Geneeskd. 2014;158:A7266 Medline.
Enns GM, Berry SA, Berry GT, Rhead WJ, Brusilow SW, Hamosh A. Survival after treatment with phenylacetate and benzoate for urea-cycle disorders. N Engl J Med. 2007;356:2282-92 Medline. doi:10.1056/NEJMoa066596
Langendonk JG, Roos JC, Angus L, et al. A series of pregnancies in women with inherited metabolic disease. J Inherit Metab Dis. 2012;35:419-24 Medline. doi:10.1007/s10545-011-9389-2
Maillot F, Cook P, Lilburn M, Lee PJ. A practical approach to maternal phenylketonuria management. J Inherit Metab Dis. 2007;30:198-201 Medline. doi:10.1007/s10545-007-0436-y
Muntau AC, Roschinger W, Habich M, et al. Tetrahydrobiopterin as an alternative treatment for mild phenylketonuria. N Engl J Med. 2002;347:2122-32 Medline. doi:10.1056/NEJMoa021654
Van der Ploeg AT, Reuser AJ. Pompe's disease. Lancet. 2008;372:1342-53 Medline. doi:10.1016/S0140-6736(08)61555-X
Van Spronsen FJ, Berger R, Smit GP, et al. Tyrosinaemia type I: orthotopic liver transplantation as the only definitive answer to a metabolic as well as an oncological problem. J Inherit Metab Dis. 1989;12(Suppl 2):339-42 Medline. doi:10.1007/BF03335416
Peters C, Steward CG. Hematopoietic cell transplantation for inherited metabolic diseases: an overview of outcomes and practice guidelines. Bone Marrow Transplant. 2003;31:229-39 Medline. doi:10.1038/sj.bmt.1703839
Artikelinformatie
Citeer dit artikel als



Reacties