
Inleiding
de ziekte
Het nagel-patellasyndroom (NPS; Mendelian Inheritance in Man, nummer 1612001, www.ncbi.nlm.nih.gov/omim/searchomim.html) is een autosomaal dominant overervende aandoening, gekenmerkt door dysplasie van de nagels, skeletafwijkingen, waaronder patella-aplasie of -hypoplasie, en frequent openkamerhoekglaucoom en nefropathie.
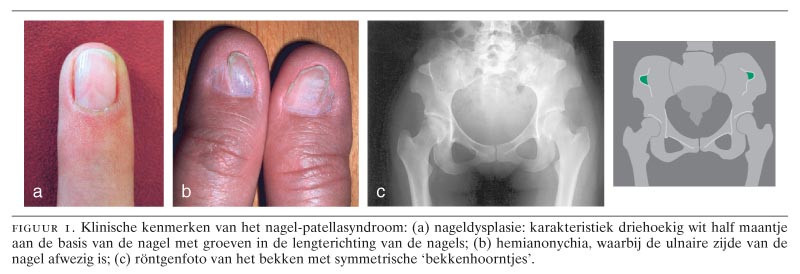
Essentiële bevindingen voor het stellen van de klinische diagnose ‘NPS’ zijn de kenmerkende afwijkingen van de dorsale structuren van de ledematen: nageldysplasie en patella-aplasie of -hypoplasie. Karakteristiek voor NPS zijn de triangulaire lunulae van de nagels (figuur 1a). Daarnaast hangt NPS samen met anonychia, hemianonychia (zie figuur 1b), groeven in de lengterichting van de nagels en gespleten nagels. De ernst van de afwijkingen van de vingernagels neemt in de anterieur-posterieure richting (van duim naar pink) af. Bij de meerderheid van de patiënten komen opvallend kleine patellae voor en in een minderheid van de gevallen zijn de patellae beiderzijds afwezig. Pathognomonisch voor NPS zijn de zogenaamde ‘bekkenhoorntjes’ (‘iliac horns’), die reeds vanaf de…
Artikelinformatie
Citeer dit artikel als




{kind=link}
Reacties