
Samenvatting
- Transporteiwitten in hepatocyten en galgangepitheelcellen zijn betrokken bij de opname en de uitscheiding van stoffen door de lever en bij de vorming van gal.
- Vele van deze eiwitten zijn recentelijk gekloneerd en gekarakteriseerd en blijken te behoren tot grote genfamilies die ook buiten de lever een rol spelen, bijvoorbeeld in het transmembraantransport in darm, nieren, placenta, longen, bloed-hersenbarrière en zaadbuizen. Zelfs in bacteriën en gisten worden verwante eiwitten gevonden. In kankercellen zijn ze betrokken bij resistentie tegen chemotherapie.
- Een aantal genetische leverziekten, zoals progressieve familiaire intrahepatische cholestase, Dubin-Johnson-syndroom, benigne recidiverende intrahepatische cholestase en zwangerschapscholestase, houdt verband met mutaties in de genen die coderen voor deze transporteiwitten.
- Ook bij door geneesmiddelen geïnduceerde cholestase en primaire biliaire cirrose zijn deze eiwitten van belang. Zo remmen ciclosporine en estradiol (glucuronide) het galzoutexporteiwit BSEP.
artikel
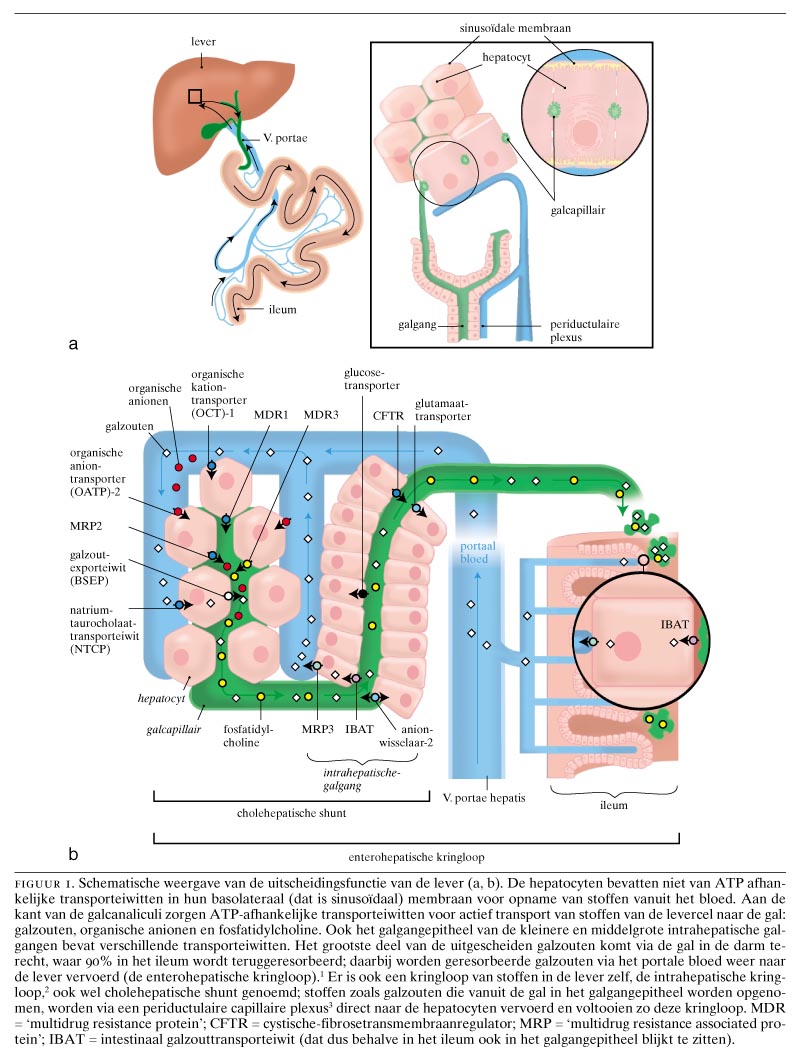
Door toepassing van moleculair-biologische technieken is het inzicht in het proces van galvorming enorm toegenomen. Transporteiwitten die hierbij een essentiële rol spelen, zijn geïdentificeerd en gekloneerd. Deze eiwitten bevinden zich zowel in de hepatocyten, het epitheel van de intrahepatische galwegen als in het terminale ileum.
De actieve uitscheiding van galzouten vanuit de hepatocyt naar de galcanaliculus vormt de belangrijkste drijvende kracht voor de vorming van gal. De gal zorgt ervoor dat bepaalde slecht in water oplosbare stoffen het lichaam via het darmkanaal kunnen verlaten. In de darm zijn galzouten van belang voor vertering en opname van voedingsvetten. Het lichaam is zuinig op zijn galzouten: in het terminale ileum worden de galzouten vanuit het darmlumen heropgenomen. Dit wordt de enterohepatische kringloop genoemd. Om deze taak uit te voeren, beschikt het terminale ileum over een galzuurtransporteiwit, de ‘ileal bile acid transporter’ (IBAT).1 Per cyclus gaat minder dan 10 van de galzouten verloren. Dit verlies wordt gecompenseerd door nieuwe aanmaak van galzouten uit cholesterol in de lever.
de galvorming
Gal wordt primair door hepatocyten gevormd. Galcanaliculi zijn microscopisch kleine kanaaltjes die door hepatocyten worden omgeven (figuur 1). Via deze kanaaltjes bereikt de gal de kleinere en grotere galkanalen om tenslotte via de extrahepatische galwegen en de galblaas in de darm te worden uitgescheiden. In tegenstelling tot de galcanaliculi hebben de intrahepatische galkanalen hun eigen epitheelbekleding: de cholangiocyten.
Transporteiwitten voor opname vanuit het poortaderbloed
Voor de opname van galzouten en andere cholefiele stoffen uit het poortaderbloed beschikt de lever over een scala aan transporteiwitten. Hiervan zijn er inmiddels ongeveer 20 gekarakteriseerd. Ze bevinden zich in het sinusoïdale membraan van de hepatocyt. Dit is het deel dat grenst aan het bloed. Het natriumafhankelijke taurocholaattransporteiwit is het belangrijkste eiwit voor de opname van galzouten.2 Het transporteiwit voor organische anionen (OATP2) is van belang voor de opname van vele stoffen in de lever en mogelijk ook voor de opname van bilirubine.4 Of deze laatste stof ook inderdaad een OATP-substraat is, moet echter nog bewezen worden. Er zijn diverse OATP's; ze bevinden zich niet alleen in de lever, maar ook in de darm, de nieren, de longen, de zaadbuizen en de bloed-hersenbarrière. Het sinusoïdale membraan van de hepatocyt bevat ook transporteiwitten voor de opname van organische kationen. Ook deze eiwitten komen in vele andere organen voor, vooral in de nieren.5 6
Transporteiwitten voor excretie in de gal
Na opname in de hepatocyt worden galzouten, bilirubine en vele andere stoffen er aan de canaliculaire zijde weer uitgepompt. Het canaliculaire membraan van de hepatocyt is hiertoe uitgerust met een reeks ATP-afhankelijke transport- of ‘pomp’-eiwitten. Het woord ‘pomp’ duidt op een actief transportproces; dit zijn actieve transporteiwitten, die stoffen tegen een 100- tot 1000-voudige concentratiegradiënt naar de gal kunnen pompen. De belangrijkste canaliculaire transporteiwitten zijn de galzoutpomp (‘bile salt export protein’ (BSEP)), de pomp voor geconjugeerd bilirubine, glutathion- en glucuronideconjugaten (‘multidrug resistance associated protein 2’ (MRP2)) en ‘multidrug resistance protein 3’ (MDR3). Dit laatste eiwit behoort evenals BSEP tot de familie van de P-glycoproteïnen en zorgt voor de uitscheiding van fosfolipiden naar de gal.7-11 Hoewel sommige van deze eiwitten vooral in de lever voorkomen, spelen soortgelijke eiwitten in bijna elk orgaan een rol bij het transport van stoffen door het celmembraan. Evolutionair gezien zijn dit belangrijke eiwitten. Gisten en bacteriën bevatten eiwitten die een aanzienlijke homologie vertonen met de transporteiwitten van zoogdieren. Resistentie van bacteriën voor antibiotica en de resistentie van kankercellen voor geneesmiddelen is voor een deel te wijten aan overexpressie van dergelijke transporteiwitten. In de lever zijn ze betrokken bij de vorming van gal.
Transporteiwitten in het galgangepitheel
Niet alleen de hepatocyten, maar ook de intrahepatische galgangepitheelcellen bevatten transporteiwitten. Het bekendst is de cystische-fibrosetransmembraanregulator (CFTR). Dit eiwit vormt een chloridekanaal dat betrokken is bij de taaislijmziekte en dat ook voorkomt in de zweetklieren, de alvleesklier, de longen, de darmen en de zaadbuizen.12 Naast het CFTR bevat het intrahepatische galgangepitheel een groot aantal andere transporteiwitten, zoals MDR1, MRP3, het reeds genoemde galzuurtransporteiwit IBAT (behalve in het ileum is dit eiwit ook in het galgangepitheel en in de niertubuli aanwezig), waterkanalen (aquaporine 1 en 4), anionuitwisselingseiwit 2 (‘anion exchanger 2’, een eiwit betrokken bij de bicarbonaatsecretie) en andere elektrolytuitwisselingseiwitten.13-16
De aanwezigheid van transporteiwitten duidt erop dat galkanalen een actieve rol spelen bij de galvorming. In dit verband dringt zich de vergelijking met het nefron op, waar in de glomerulus de primaire urine wordt gevormd terwijl in de proximale en distale tubuli de urine wordt geconcentreerd, waarbij tevens allerlei stoffen worden geresorbeerd en andere worden uitgescheiden. Zo zou men in de lever kunnen spreken van primaire gal, die door de hepatocyten wordt gemaakt. In de galkanaaltjes verandert de galsamenstelling door toedoen van transporteiwitten. Dit laatste heeft waarschijnlijk te maken met het feit dat in de gal sprake is van een precaire balans tussen elkaar in oplossing houdend cholesterol, fosfolipiden en galzouten. Als het evenwicht verstoord is, slaat cholesterol neer in de vorm van galstenen. De transportfunctie van de galkanaaltjes dient ertoe om nuttige stoffen, zoals bepaalde aminozuren en galzouten, vanuit de gal terug te resorberen zonder dit evenwicht te verstoren. Daarnaast zorgt de intrahepatische kringloop van deze osmotisch actieve stoffen voor een extra nettoflux van water.
Leverstamcellen
Tussen de hepatocyten en de intrahepatische galgangen bevinden zich de kanalen van Hering.17 Deze kanalen zijn deels door hepatocyten en deels door galgangepitheel omgeven. Hier bevinden zich ook de ‘ovale cellen’, die wel als de stamcellen van de lever worden beschouwd. Deze cellen kunnen prolifereren en zijn pluripotent: zij kunnen uitgroeien tot zowel hepatocyten als cholangiocyten. Zolang bij ernstige leverschade het stamcelcompartiment onaangetast blijft, kan de lever regenereren.18 19 De stamcellen vertonen een sterke expressie van MDR1, MRP1 en MRP3.20 Deze transporteiwitten hebben hier waarschijnlijk een beschermende functie, omdat zij hepatotoxische stoffen de stamcel uit kunnen pompen.
progressieve familiaire intrahepatische cholestase type 1, 2 en 3
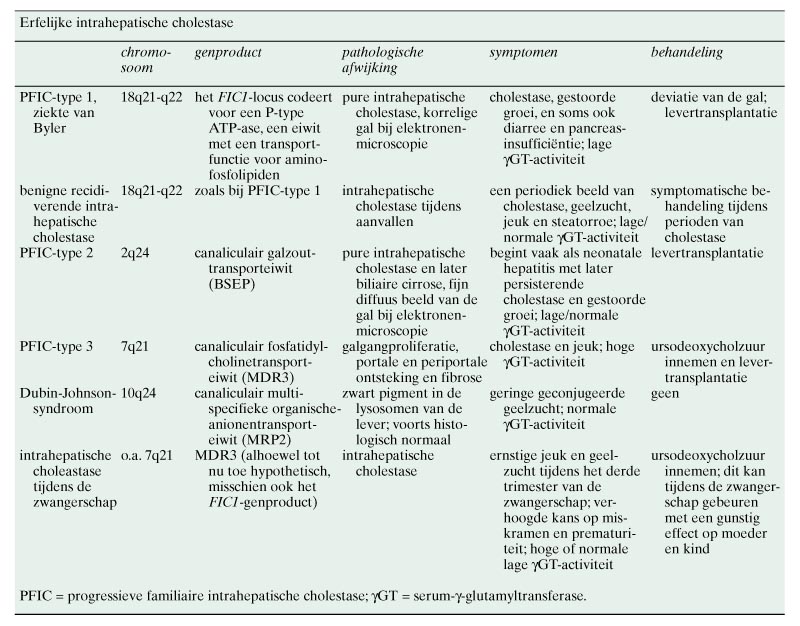
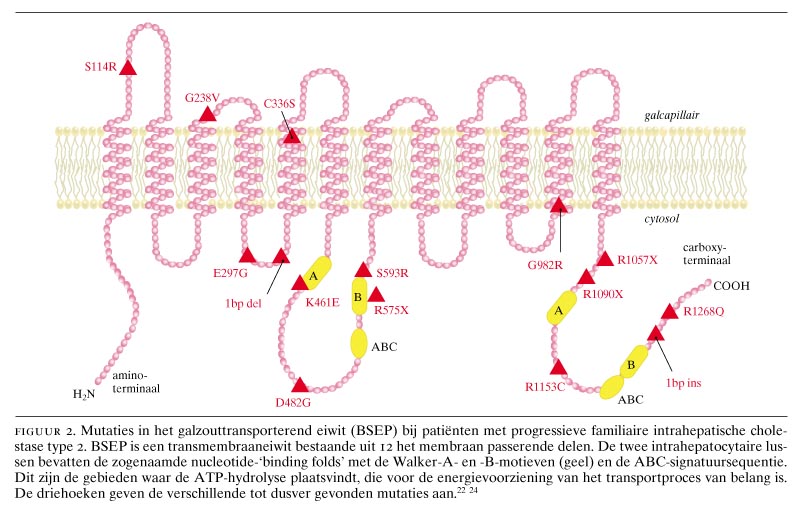
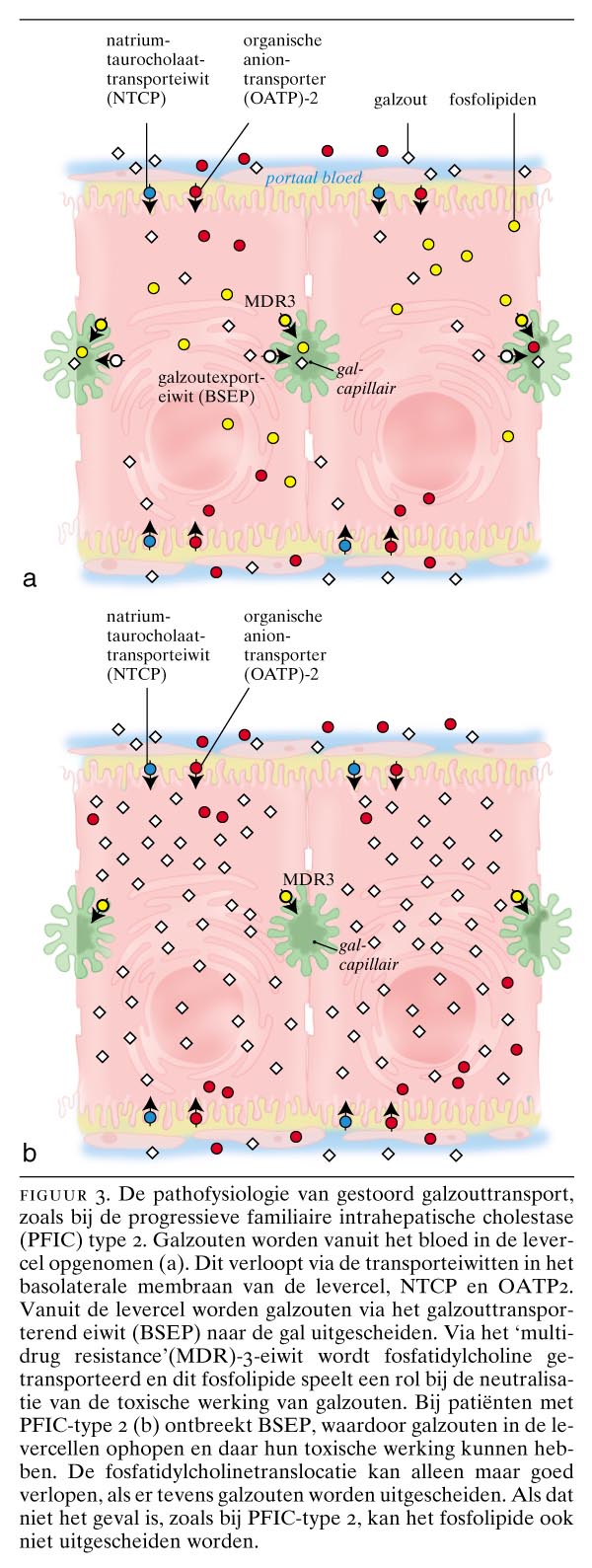
Genetische afwijkingen zijn spelingen van de natuur die inzicht kunnen verschaffen in de functie van een bepaald eiwit. Toen bekend werd dat BSEP het galzouttransporteiwit van de lever was, deed zich de vraag voor wat het fenotype van de ziekte zou zijn waarbij BSEP gemuteerd is. De aandacht richtte zich toen op congenitale cholestatische ziekten. Genetisch onderzoek had aangetoond dat het gen voor de ziekte van Byler, een vorm van congenitale cholestase die vooral, maar niet uitsluitend, bij de Pennsylvanian Dutch voorkomt, op chromosoom 18q21 ligt.21 Dit kon niet het gen voor BSEP zijn, want hiervan was inmiddels gebleken dat het op chromosoom 2q24 ligt. Ook was duidelijk dat er naast de ziekte van Byler nog tenminste 2 andere groepen patiënten met erfelijke cholestase waren. Voor één van deze groepen bleek het gen op chromosoom 2q24 te liggen.22 Strautnieks et al. hebben aangetoond dat deze patiënten een mutatie van het gen voor BSEP hebben,23 en wij hebben vervolgens gevonden dat het BSEP-eiwit hierbij niet tot expressie komt.24 Deze aandoening, die progressieve familiaire intrahepatische cholestase type 2 (PFIC-type 2) wordt genoemd, is de belangrijkste van de groep van familiaire intrahepatische cholestasesyndromen, waar inmiddels 3 subtypen van beschreven zijn. Deze zijn in de tabel vermeld. Karakteristiek voor de ziekte van Byler (PFIC-type 1) en voor PFIC-type 2 is een lage serumactiviteit van ?-glutamyltransferase (?GT), en dit ondanks de cholestase en de daarbijbehorende verhoogde alkalische-fosfataseactiviteit. De tot nu toe gevonden mutaties voor BSEP staan in figuur 2 afgebeeld. Figuur 3 geeft een schematische indruk van de pathofysiologische gevolgen van BSEP-deficiëntie bij patiënten met PFIC-type 2.
Het MDR3-gen codeert voor een canaliculair fosfatidylcholinetransporterend eiwit. Fosfatidylcholine is het belangrijkste fosfolipide in de gal en speelt een rol bij het in oplossing houden van cholesterol en het neutraliseren van de toxische werking van galzouten. Bij afwezigheid van het MDR3-eiwit, zoals dat het geval is bij PFIC-type 3, ontstaat er in de lever een beeld van ernstige galgangschade met galgangproliferatie en portale en periportale ontsteking en fibrose. PFIC-type 3 gaat gepaard met een hoge serumactiviteit van ?GT, hetgeen duidt op beschadiging van de intrahepatische galgangen waarin dit enzym voornamelijk gelokaliseerd is.25 26
benigne recidiverende intrahepatische cholestase en intrahepatische cholestase bij zwangerschap
Benigne recidiverende intrahepatische cholestase (BRIC) en intrahepatische cholestase tijdens de zwangerschap zijn aandoeningen waarbij de genoemde genen ook betrokken zijn. BRIC is een aandoening die gepaard gaat met perioden van geelzucht, jeuk en steatorroe. Tussen de aanvallen door is de leverfunctie normaal. Het gen voor deze ziekte ligt in hetzelfde locus als dat voor PFIC-type 1.27-29 Het lijkt dus waarschijnlijk dat waar bij PFIC-type 1 sprake is van een volledig gendefect, het bij BRIC gaat om een afwijkende regulatie van genexpressie. De rol van het bij deze aandoeningen betrokken gen, FIC1, is echter nog niet volledig opgehelderd.
In families met PFIC-type 3 komt de intrahepatische cholestase tijdens de zwangerschap met een verhoogde frequentie voor.30 31 Evenals dit bij PFIC-type 3 het geval is, hebben deze patiënten tijdens de periode van cholestase in het 3e trimester van de zwangerschap een verhoogde serumactiviteit van ?GT. Een aantal van deze patiënten blijkt heterozygoot voor de mutatie van het MDR3-gen te zijn. Dit is echter niet het einde van dit verhaal. Er zijn ook patiënten met een normale serum-?GT-activiteit. Mogelijk houdt deze vorm van zwangerschapscholestase verband met BRIC, want ook in families van BRIC-patiënten komt deze ziekte vaker voor.32
primaire biliaire cirrose
Primaire biliaire cirrose vertoont histologisch enige gelijkenis met het beeld bij PFIC-type 3. Bij primaire biliaire cirrose is er echter tevens een auto-immune ontregeling met vorming van antimitochondriale antistoffen. Het is niet uitgesloten dat galzouten betrokken zijn bij deze ontregeling. Mitochondriën zijn gevoelig voor galzouten en uit immunohistologisch onderzoek blijkt dat de autoreactiviteit zich voornamelijk richt tegen het mitochondriale pyruvaatdehydrogenase. Dus mogelijk zijn de antimitochondriale antistoffen het gevolg van een door galzouten geïnduceerde beschadiging van het galgangepitheel. Daarnaast zijn waarschijnlijk omgevingsinvloeden bij primaire biliaire cirrose van belang, hetgeen blijkt uit de zeer hoge prevalentie van de ziekte in sterk geïndustrialiseerde gebieden, zoals de omgeving van Newcastle upon Tyne.33-35 Wellicht leiden omgevingsinvloeden tot een ontregeling van MDR3-gemedieerd transport met galgangbeschadiging en stimulatie van autoreactiviteit tot gevolg.
dubin-johnson-syndroom
Het Dubin-Johnson-syndroom is een onschuldige aandoening, die gepaard gaat met een geconjugeerde hyperbilirubinemie. Inzicht in dit syndroom heeft een belangrijke stimulans gekregen door de ontdekking van de zogenaamde transportdeficiënte rat.36 Dit proefdier heeft een op het Dubin-Johnson-syndroom gelijkende aandoening als gevolg van een mutatie van het MRP2-gen;37 38 daardoor is er geen functioneel MRP2. Patiënten met het Dubin-Johnson-syndroom hebben een normale levensverwachting en zijn asymptomatisch, met uitzondering van een gele huid en ogen. Deze aandoening is net als bij de transportdeficiënte rat het gevolg van een mutatie van het bilirubine-glucuronidetransporteiwit MRP2 in het canaliculaire membraan van de lever.39-41 De verhoogde concentratie van geconjugeerd bilirubine in het bloed is het gevolg van een compensatoire toename van het MRP3-eiwit in het basolaterale membraan van de hepatocyt.42 43 Onder normale omstandigheden komt dit eiwit niet tot expressie in de lever, maar bij het Dubin-Johnson-syndroom wel. In tegenstelling tot MRP2 transporteert MRP3 bilirubineconjugaten van lever naar bloed. Histologisch is de lever bij patiënten met het Dubin-Johnson-syndroom normaal, met uitzondering van een karakteristiek zwart lysosomaal pigment.44
door geneesmiddelen geïnduceerde cholestase
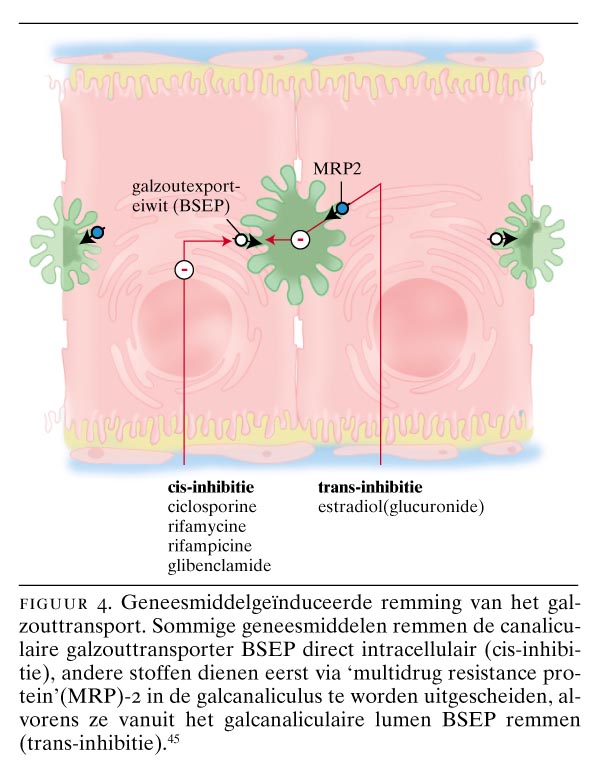
Door geneesmiddelen geïnduceerde cholestase is voor een deel het gevolg van de interactie van geneesmiddelen met transporteiwitten in de lever. Hierbij zijn vooral het MRP2 en het BSEP van belang. BSEP kan zowel vanaf de cellulaire kant geremd worden (zogenaamde cis-inhibitie), als vanaf de canaliculaire kant (trans-inhibitie).45 Ciclosporine behoort tot de eerste categorie en estradiol (glucuronide) tot de tweede categorie van BSEP-remmers (figuur 4). Estradiol (glucuronide) wordt eerst via MRP2 in de gal uitgescheiden om vervolgens BSEP vanuit het canaliculaire lumen te remmen. In transportdeficiënte ratten die niet over MRP2 beschikken, veroorzaakt estradiol (glucuronide) dan ook geen cholestase.
Ook cytokinen kunnen de expressie van canaliculaire transporteiwitten beïnvloeden.46 Deze beïnvloeding vindt gedeeltelijk plaats op transcriptieniveau. Bij sommige vormen van virale hepatitis is er mogelijk sprake van een door cytokinen gemedieerde verminderde expressie van MRP2 en BSEP.
conclusie
Moleculair-genetisch en celbiologisch onderzoek heeft geleid tot een beter begrip van het proces van galvorming en tot identificatie van een aantal genetisch-cholestatische ziekten. Vanuit deze kennis is het wellicht mogelijk om ook complexere leverziekten te begrijpen en zo aanknopingspunten te vinden voor nieuwe behandelingsmogelijkheden.
Het onderzoek dat aan dit artikel ten grondslag ligt, vindt plaats in een samenwerkingsverband met de afdelingen Pathologie en Laboratoriumgeneeskunde (hoofd: prof.dr.S.Poppema), Medische Oncologie (hoofd: mw.prof.dr.E.G.E.de Vries), Hepatobiliaire Chirurgie (hoofd: prof.dr.M.J.H.Slooff), Farmacokinetiek en Drug Delivery (hoofd: prof.dr.D.K.F.Meijer), Medische Genetica (hoofd: prof.dr.C.H.C.M.Buys) en het Centraal Dierenlaboratorium. Het werk wordt financieel ondersteund door de Rijksuniversiteit Groningen, het Academisch Ziekenhuis Groningen, de Nederlandse Organisatie voor Wetenschappelijk Onderzoek NWO, de De Cock Stichting, de Maag-, Darm- en Leverstichting, Tramedico BV, Astra-Zeneca Nederland BV en Byk-Gulden Nederland BV.
Literatuur
Craddock AL, Love MW, Daniel RW, Kirby LC, Walters HC,Wong MH, et al. Expression and transport properties of the human ileal andrenal sodium-dependent bile acid transporter. Am J Physiol1998;274:G157-69.
Jacquemin E, Hagenbuch B, Stieger B, Wolkoff AW, Meier PJ.Expression cloning of a rat liver Na(+)-independent organic aniontransporter. Proc Natl Acad Sci U S A 1994;91:133-7.
Ekataksin W, Wake K. New concepts in biliary and vascularanatomy of the liver. Prog Liver Dis 1997;15:1-30.
Reichel C, Gao B, Montfoort J van, Cattori V, Rahner C,Hagenbuch B, et al. Localization and function of the organicanion-transporting polypeptide Oatp2 in rat liver. Gastroenterology1999;117:688-95.
Koepsell H. Organic cation transporters in intestine,kidney, liver, and brain. Annu Rev Physiol 1998;60:243-66.
Koepsell H, Gorboulev V, Arndt P. Molecular pharmacologyof organic cation transporters in kidney. J Membr Biol1999;167:103-17.
Gerloff T, Stieger B, Hagenbuch B, Madon J, Landmann L,Roth J, et al. The sister of P-glycoprotein represents the canalicular bilesalt export pump of mammalian liver. J Biol Chem 1998;273:10046-50.
Konig J, Nies AT, Cui Y, Leier I, Keppler D. Conjugateexport pumps of the multidrug resistance protein (MRP) family: localization,substrate specificity, and MRP2-mediated drug resistance. Biochim BiophysActa 1999;1461:377-94.
Kamisako T, Leier I, Cui Y, Konig J, Buchholz U,Hummel-Eisenbeiss J, et al. Transport of monoglucuronosyl and bisglucuronosylbilirubin by recombinant human and rat multidrug resistance protein 2.Hepatology 1999;30:485-90.
Smith AJ, Vree JM de, Ottenhoff R, Oude Elferink RP,Schinkel AH, Borst P. Hepatocyte-specific expression of the human MDR3P-glycoprotein gene restores the biliary phosphatidylcholine excretion absentin Mdr2 (-/-) mice. Hepatology 1998;28:530-6.
Smith AJ, Timmermans-Hereijgers JL, Roelofsen B, WirtzKW, Blitterswijk WJ van, Smit JJ, et al. The human MDR3 P-glycoproteinpromotes translocation of phosphatidylcholine through the plasma membrane offibroblasts from transgenic mice. FEBS Lett 1994;354:263-6.
Riordan JR, Chang XB. CFTR, a channel with the structureof a transporter. Biochim Biophys Acta 1992;1101:221-2.
Medina JF, Martinez-Anso, Vazquez JJ, Prieto J. Decreasedanion exchanger 2 immunoreactivity in the liver of patients with primarybiliary cirrhosis. Hepatology 1997;25:12-7.
Lazaridis KN, Pham LD, Tietz P, Marinelli RA, Groen PCde, Levine S, et al. Rat cholangiocytes absorb bile acids at their apicaldomain via the ileal sodium-dependent bile acid transporter. J Clin Invest1997;100:2714-21.
Lazaridis KN, Pham LD, Vroman B, Groen PC de, LaRusso NF.Kinetic and molecular identification of sodium-dependent glucose transporterin normal rat cholangiocytes. Am J Physiol 1997; 272:G1168-74.
Marinelli RA, Tietz PS, Pham LD, Rueckert L, Agre P,LaRusso NF. Secretin induces the apical insertion of aquaporin-1 waterchannels in rat cholangiocytes. Am J Physiol 1999;276:G280-6.
Theise ND, Saxena R, Portmann BC, Thung SN, Yee H,Chiriboga L, et al. The canals of Hering and hepatic stem cells in humans.Hepatology 1999;30:1425-33.
Fausto N. Liver regeneration. J Hepatol 2000;32(1Suppl):19-31.
Thorgeirsson SS. Hepatic stem cells in liverregeneration. FASEB J 1996;10:1249-56.
Ros JE, Geuken M, LaRusso NF, Thorgeirsson SS, JansenPLM, Müller M. ABC-transporter gene expression in cholangiocytes andhepatic oval cells reveals a multidrug resistance phenotype. Hepatology1999;30:432A.
Bull LN, Carlton VE, Stricker NL, Baharloo S, DeYoung JA,Freimer NB, et al. Genetic and morphological findings in progressive familialintrahepatic cholestasis (Byler disease PFIC-1 and Bylersyndrome): evidence for heterogeneity. Hepatology 1997; 26:155-64.
Strautnieks SS, Bull LN, Knisely AS, Kocoshis SA, Dahl N,Arnell H, et al. A gene encoding a liver-specific ABC transporter is mutatedin progressive familial intrahepatic cholestasis. Nat Genet 1998;20:233-8.
Strautnieks SS, Kagalwalla AF, Tanner MS, Gardiner RM,Thompson RJ. Locus heterogeneity in progressive familial intrahepaticcholestasis. J Med Genet 1996;33:833-6.
Jansen PL, Strautnieks SS, Jacquemin E, Hadchouel M,Sokal EM, Hooiveld GJ, et al. Hepatocanalicular bile salt export pumpdeficiency in patients with progressive familial intrahepatic cholestasis.Gastroenterology 1999;117:1370-9.
Vree JM de, Jacquemin E, Sturm E, Cresteil D, Bosma PJ,Aten J, et al. Mutations in the MDR3 gene cause progressive familial intrahepatic cholestasis. Proc Natl Acad Sci U S A 1998;95:282-7.
Deleuze JF, Jacquemin E, Dubuisson C, Cresteil D, DumontM, Erlinger S, et al. Defect of multidrug-resistance 3 gene expression in asubtype of progressive familial intrahepatic cholestasis. Hepatology1996;23:904-8.
Bull LN, Eijk MJ van, Pawlikowska L, DeYoung JA, JuijnJA, Liao M, et al. A gene encoding a P-type ATPase mutated in two forms ofhereditary cholestasis. Nat Genet 1998;18:219-24.
Sinke RJ, Carlton VE, Juijn JA, Delhaas T, Bull L, BergeHenegouwen GP van, et al. Benign recurrent intrahepatic cholestasis (BRIC):evidence of genetic heterogeneity and delimitation of the BRIC locus to a7-cM interval between D18S69 and D18S64. Hum Genet 1997;100:382-7.
Bull LN, Juijn JA, Liao M, Eijk MJ van, Sinke RJ,Stricker NL, et al. Fine-resolution mapping by haplotype evaluation: theexamples of PFIC1 and BRIC. Hum Genet 1999;104:241-8.
Jacquemin E, Cresteil D, Manouvrier S, Boute O, HadchouelM. Heterozygous non-sense mutation of the MDR3 gene in familial intrahepaticcholestasis of pregnancy letter. Lancet 1999;353:210-1.
Dixon PH, Weerasekera N, Linton KJ, Donaldson O, ChambersJ, Egginton E, et al. Heterozygous MDR3 missense mutation associated withintrahepatic cholestasis of pregnancy: evidence for a defect in proteintrafficking. Hum Mol Genet 2000;9:1209-17.
Pagter AG de, Berge Henegouwen GP van, Bokkel Huinink JAten, Brandt KH. Familial benign recurrent intrahepatic cholestasis.Interrelation with intrahepatic cholestasis of pregnancy and from oralcontraceptives? Gastroenterology 1976;71:202-7.
Howel D, Fischbacher CM, Bhopal RS, Gray J, Metcalf JV,James OF. An exploratory population-based case-control study of primarybiliary cirrhosis. Hepatology 2000;31:1055-60.
James OF, Bhopal RS, Howel D, Gray J, Burt AD, MetcalfJV. Primary biliary cirrhosis once rare, now common in the United Kingdom?Hepatology 1999;30:390-4.
Jones DE, Watt FE, Metcalf JV, Bassendine MF, James OF.Familial primary biliary cirrhosis reassessed: a geographically-basedpopulation study. J Hepatol 1999;30:402-7.
Jansen PL, Peters WH, Lamers WH. Hereditary chronicconjugated hyperbilirubinemia in mutant rats caused by defective hepaticanion transport. Hepatology 1985;5:573-9.
Paulusma CC, Bosma PJ, Zaman GJ, Bakker CT, Otter M,Scheffer GL, et al. Congenital jaundice in rats with a mutation in amultidrug resistance-associated protein gene. Science1996;271:1126-8.
Buchler M, Konig J, Brom M, Kartenbeck J, Spring H, HorieT, et al. cDNA cloning of the hepatocyte canalicular isoform of the multidrugresistance protein, cMrp, reveals a novel conjugate export pump deficient inhyperbilirubinemic mutant rats. J Biol Chem 1996;271:15091-8.
Paulusma CC, Kool M, Bosma PJ, Scheffer GL, Borg F ter,Scheper RJ, et al. A mutation in the human canalicular multispecific organicanion transporter gene causes the Dubin-Johnson syndrome. Hepatology1997;25:1539-42.
Kartenbeck J, Leuschner U, Mayer R, Keppler D. Absence ofthe canalicular isoform of the MRP gene-encoded conjugate export pump fromthe hepatocytes in Dubin-Johnson syndrome. Hepatology1996;23:1061-6.
Toh S, Wada M, Uchiumi T, Inokuchi A, Makino Y, Horie Y,et al. Genomic structure of the canalicular multispecific organicanion-transporter gene (MRP2/cMOAT) and mutations in the ATP-binding-cassetteregion in Dubin-Johnson syndrome. Am J Hum Genet 1999;64:739-46.
Hirohashi T, Suzuki H, Ito K, Ogawa K, Kume K, Shimizu T,et al. Hepatic expression of multidrug resistance-associated protein-likeproteins maintained in eisai hyperbilirubinemic rats. Mol Pharmacol1998;53:1068-75.
Hirohashi T, Suzuki H, Sugiyama Y. Characterization ofthe transport properties of cloned rat multidrug resistance-associatedprotein 3 (MRP3). J Biol Chem 1999;274:15181-5.
Kitamura T, Alroy J, Gatmaitan Z, Inoue M, Mikami T,Jansen P, et al. Defective biliary excretion of epinephrine metabolites inmutant (TR-) rats: relation to the pathogenesis of black liver in theDubin-Johnson syndrome and Corriedale sheep with an analogous excretorydefect. Hepatology 1992;15:1154-9.
Stieger B, Fattinger K, Madon J, Kullak-Ublick GA, MeierPJ. Drug- and estrogen-induced cholestasis through inhibition of thehepatocellular bile salt export pump (Bsep) of rat liver. Gastroenterology2000;118:422-30.
Vos TA, Hooiveld GJ, Koning H, Childs S, Meijer DK,Moshage H, et al. Up-regulation of the multidrug resistance genes, Mrp1 andMdr1b, and down-regulation of the organic anion transporter, Mrp2, and thebile salt transporter, Spgp, in endotoxemic rat liver. Hepatology1998;28:1637-44.
Artikelinformatie
Citeer dit artikel als




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Reacties