
Dames en Heren,
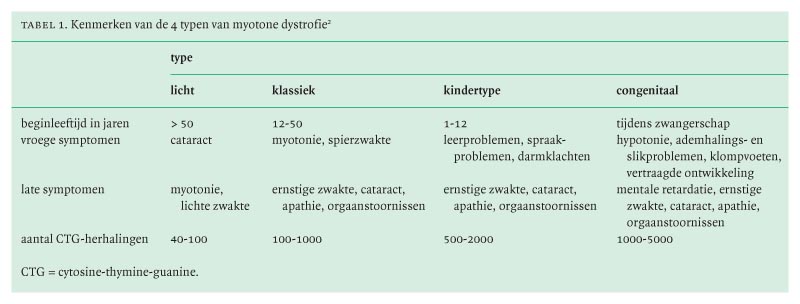
Myotone dystrofie is de frequentste erfelijke neuromusculaire aandoening. De ziekte heet ook wel de ziekte van Curschmann-Steinert en erft autosomaal dominant over.1 Men onderscheidt 4 typen myotone dystrofie: het lichte (Engels: ‘mild’) type, het klassieke of volwassen type, het kindertype en het congenitale type ofwel congenitale myotone dystrofie (tabel 1).2 Dit congenitale type komt bij 1:3500-1:16.000 pasgeborenen voor en kent een hoge sterfte en morbiditeit.3 4 Transmissie van congenitale myotone dystrofie vindt zelden via de vader en meestal via de moeder plaats, maar deze is zich meestal niet van haar ziekte bewust.5 Omdat prenatale diagnostiek en erfelijkheidsadvies mogelijk zijn, is het belangrijk zwangeren met myotone dystrofie tijdig te herkennen. Dit geldt voor zowel de huisarts, de gynaecoloog als de verloskundige. Zij moeten bekend zijn met de symptomen die kunnen wijzen op congenitale myotone dystrofie bij de foetus. In deze les bespreken…
Artikelinformatie
Citeer dit artikel als



{kind=link}
Reacties