
artikel
Dames en Heren,
Familiaire Middellandse Zeekoorts (‘familial Mediterranean fever’, FMF) is een aangeboren, steriele, aanvalsgewijze ontstekingsziekte. Onder de autochtone Nederlandse bevolking is FMF extreem zeldzaam, maar deze erfelijke aandoening komt veel voor bij migranten uit het Middellandse Zeegebied. Vroege herkenning maakt een tijdige behandeling met colchicine mogelijk. Als dit niet gebeurt, kunnen ernstige complicaties optreden, zoals terminale nierinsufficiëntie. Aan de hand van 3 casussen laten we zien dat de herkenning van FMF lastig kan zijn.
Patiënt A, de 1e dochter van gezonde, niet-verwante Marokkaanse ouders, was 3 jaar oud toen haar ouders haar naar de kinderarts brachten wegens heftige buikpijn. Zij was die nacht door de pijn wakker geworden. Ze had hoge koorts en wilde niet meer eten; ze braakte niet. Patiënte had dagelijks zachte ontlasting gehad en dat patroon was niet veranderd. De ouders herkenden het beeld: al vanaf de leeftijd van 14 maanden had patiënte elke 4-8 weken een dergelijke aanval. Het begin was steeds acuut met pijn en eetlustverlies, en ze had meestal ook koorts. Dit hield 2-3 dagen aan, waarna patiënte snel en volledig opknapte.
Bij lichamelijk onderzoek zagen wij een matig ziek meisje met een temperatuur van 38,5ºC. Bij palpatie van de buik was er linksonder spierverzet en drukpijn. Bij bloedonderzoek was de CRP-waarde 40 mg/l; er was geen leukocytose. De urine bevatte geen leukocyten of eiwit.
Op grond van de terugkerende buikpijn, de koorts, de verhoogde CRP-waarde en de Marokkaanse achtergrond dacht de kinderarts aan FMF. Om de werkdiagnose te toetsen schreef zij colchicine 0,5 mg 1 dd voor, waarna de aanvallen verminderden. Toch kreeg patiënte eens in de 4 maanden koorts, buikpijn en eenzijdige thoracale pijn, die paste bij pleuritis. Na het ophogen van de dosering naar 0,5 mg 2 dd bleven de aanvallen uiteindelijk weg. DNA-analyse toonde in beide allelen van het MEFV-gen dezelfde mutatie: p.M694I, waarbij het aminozuur methionine op positie 694 in het eiwit is vervangen door isoleucine.
Patiënt B, het 3e kind van niet-verwante Turkse ouders, kreeg vanaf de leeftijd van 1,5 jaar aanvallen van hoge koorts en buikpijn. Deze begonnen abrupt en vaak kreeg patiënte dan koude rillingen. De aanvallen duurden kort, enige uren tot hooguit 2 dagen, en gingen gepaard met hoofdpijn en soms met retrosternale pijn of pijn in de flank. Pas toen ze 8 jaar was, werd de diagnose ‘FMF’ gesteld. DNA-analyse toonde een samengestelde heterozygotie van het MEFV-gen. De mutaties waren p.M694I en p.M694V; bij de 2e variant is methionine vervangen door valine.
Aanvankelijk was behandeling met colchicine 0,5 mg 2 dd effectief, maar toen ze 10 jaar oud was kwamen de aanvallen zeer frequent terug en kreeg ze dikwijls ook monoartritis van de enkel of knie. Verhoging van de dagelijkse colchicinedosering naar 1 mg 2 dd en toevoeging van ibuprofen 10 mg/kg 3dd bracht geen verbetering. Bij bloedonderzoek waren de CRP- en BSE-waarden steeds sterk verhoogd. Ook tijdens een opname met gesuperviseerde medicatie-inname ontwikkelde zij koorts, buikpijn en acute monoartritis van de knie. Hierop startten wij behandeling met recombinant humaan interleukine(IL)-1-receptorantagonist anakinra 100 mg 1 dd, naast colchicine. Binnen enkele dagen verdwenen de ontstekingsverschijnselen en na 4 dagen was de CRP-waarde genormaliseerd. Onder behandeling met anakinra en colchicine bleef de ziekte hierna jarenlang in remissie. Pogingen de anakinra te staken werden tot 3 maal toe gevolgd door een exacerbatie binnen 36 h. De medicatie werd daarna niet meer gewijzigd en de ziekte bleef in remissie op zeer incidentele aanvallen na.
Patiënt C was een vrouw die op 18-jarige leeftijd uit Oost-Turkije naar Nederland was gekomen. Vanaf haar 14e jaar had zij recidiverende aanvallen van buikpijn, die soms gepaard gingen met koorts. De aanvallen gingen steeds binnen 5 dagen spontaan over. Vanwege deze klachten werd zij enkele malen ter observatie opgenomen, maar een duidelijke diagnose kon niet worden gesteld, ondanks consultatie van de chirurg, gynaecoloog en internist. De pijn werd uiteindelijk geweten aan prikkelbaredarmsyndroom, ook al werden er herhaaldelijk verhoogde waarden van BSE (58-116 mm/1e h) en CRP (15-229 mg/l) gevonden.
Op de leeftijd van 34 jaar ontwikkelde patiënte een nefrotisch syndroom. Uit een nierbiopsie bleek dat dit kwam door AA-amyloïdose. Hierop werd de diagnose ‘FMF’ klinisch gesteld en met genetisch onderzoek ondersteund. In beide allelen van het MEFV-gen werd dezelfde mutatie gevonden: p.M680I. We behandelden patiënte met colchicine 0,5 mg 3 dd, waarna de buikpijnaanvallen verdwenen. De nierschade schreed echter voort en op de leeftijd van 37 jaar ontwikkelde zij terminale nierinsufficiëntie. Patiënte kreeg 6 maanden na het starten van de hemodialyse een niertransplantaat met een donornier afkomstig van haar moeder. Een half jaar na de transplantatie maakte zij het goed. Haar creatinineklaring bedroeg 45 ml/min en de FMF was met colchicine 0,5 mg 3 dd in remissie.
Beschouwing
Wij beschreven 3 patiënten van Turkse of Marokkaanse afkomst, die gediagnosticeerd werden met familiaire Middellandse Zeekoorts.
Epidemiologie
FMF komt bij uitstek voor bij Turken, Armeniërs, oriëntaalse Joden en Arabieren, inclusief Noord-Afrikanen; bij andere bevolkingsgroepen, zoals autochtone Nederlanders, is de ziekte extreem zeldzaam. In delen van Noordoost-Turkije en Armenië is de prevalentie van FMF zeer hoog, namelijk 1:500.1 Voor heel Turkije wordt de prevalentie geschat op 1:1000. De prevalentie in Marokko is onbekend. In Nederland wonen meer dan 800.000 migranten uit gebieden waar FMF endemisch is. Daarom zijn hier vermoedelijk honderden patiënten met FMF. Hoe vaak de ziekte werkelijk in ons land voorkomt, is echter onbekend.
Klinisch beeld
FMF is een erfelijke auto-inflammatoire ziekte. Het kenmerk van auto-inflammatoire ziekten is een chronische of recidiverende ontsteking zonder infectie of auto-immuniteit. Bij FMF komt de ontsteking op in aanvallen die gemiddeld 12-72 h duren. Tijdens de aanvallen hebben patiënten meestal koorts (96%) en gelokaliseerde ontstekingsverschijnselen, vooral serositis en artritis.2 De serositis kan alle lichaamsvliezen treffen in de vorm van pleuritis, pericarditis, peritonitis of, bij jongens en mannen, periorchitis. Aanvallen met peritonitis komen bij > 90% van de patiënten voor. Vaak leidt dit tot nodeloos chirurgisch ingrijpen. Artritis komt bij 45-75% van de patiënten voor, is meestal acuut en beperkt zich tot 1 of enkele van de grote gewrichten.
Veel patiënten hebben spierpijn tijdens de aanvallen. Een typische huidafwijking is erysipelasachtig erytheem van de enkelregio (figuur); dit komt voor bij 10-40% van de patiënten.2 Na een acute koortsaanval kunnen patiënten soms last hebben van aanhoudende spierpijn en in zeldzame gevallen van chronische monoartritis. De meeste patiënten zijn tussen de koortsaanvallen echter weken tot maanden lang volledig klachtenvrij. De duur van de klinische remissies wisselt in de loop der tijd en verschilt tussen patiënten onderling.
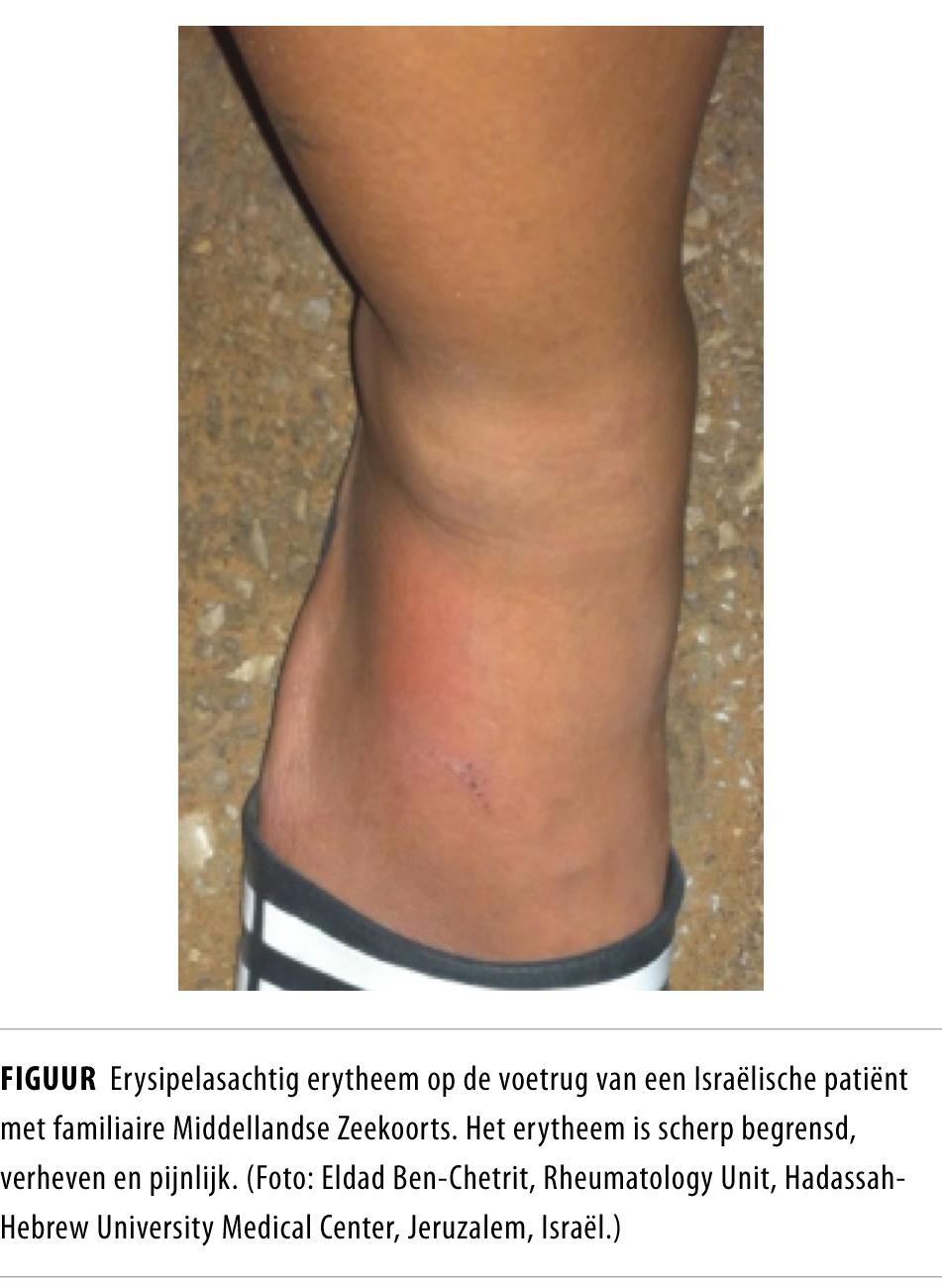
Het ontstekingsproces kan tussen de aanvallen subklinisch doorgaan. Hierin schuilt het grootste gevaar van FMF.3 Wanneer de concentraties van ontstekingseiwitten langdurig verhoogd zijn, kunnen deze eiwitten, met name het serumamyloïd A (SAA), in de loop der jaren als amyloïd neerslaan in organen; dit gebeurt vooral in de nieren. Deze renale AA-amyloïdose leidt tot een nefrotisch syndroom en uiteindelijk tot terminale nierinsufficiëntie, zoals bij patiënt C. In zeldzame gevallen is amyloïdose zelfs de eerste presentatie van FMF; bij deze patiënten is de voorafgaande ontsteking niet herkend of is de ontsteking geheel subklinisch verlopen.
Bij 80% van de patiënten beginnen de klachten op de kinderleeftijd. De eerste ziekteverschijnselen kunnen al op de zuigelingenleeftijd optreden, maar zich ook pas tijdens de vroege volwassenheid openbaren.
Diagnose
Herhaalde koortsaanvallen zonder focus die gepaard gaan met hoge waarden van ontstekingsparameters en die worden afgewisseld met volledig klachtenvrije intervallen gedurende weken tot maanden zijn verdacht voor een periodiek koortssyndroom. Vooral bij patiënten van mediterrane afkomst moet dit beeld doen denken aan FMF. Bij een dergelijk beloop zijn een infectie, maligniteit of auto-immuunziekte minder waarschijnlijk. Bij jonge kinderen zijn infecties echter zo gebruikelijk dat het vaak jaren duurt voordat een arts aan FMF denkt, zelfs in landen met een hoge prevalentie van de ziekte. Een late diagnose zoals bij onze patiënten is geen uitzondering, vooral omdat het centrale symptoom, koorts, soms ontbreekt.
De diagnose berust op klinische criteria (tabel).4 Wanneer een patiënt van mediterrane afkomst voldoet aan 2 majeure criteria of aan 1 majeur criterium en 2 mineure criteria, heeft hij of zij zeer waarschijnlijk FMF. DNA-onderzoek kan de diagnose wel meer of minder waarschijnlijk maken, maar niet bewijzen of verwerpen.

Pathogenese
De reden dat DNA-onderzoek de diagnose niet kan bewijzen of verwerpen heeft te maken met de complexe genetica van FMF. De ziekte erft namelijk meestal, maar niet altijd autosomaal recessief over. In 1997 is het verantwoordelijke gen MEFV (dit staat voor MEditerranean FeVer) geïdentificeerd. Het komt tot expressie in fagocyten. Nu, 15 jaar later, is de functie van het genproduct van MEFV, pyrine, nog steeds niet precies bekend. Waarschijnlijk is pyrine een bouwsteen van inflammasomen. Dit zijn cytoplasmatische eiwitcomplexen die het pro-inflammatoire cytokine interleukine-1β activeren. De onderzochte mutaties in MEFV leiden tot een versterkte functie van pyrine en tot een verhoogde productie van IL-1β.5 Dit heeft belangrijke therapeutische consequenties, zoals blijkt uit de ziektegeschiedenis van patiënt B. Haar ziekte kwam in remissie door behandeling met anakinra, een specifieke remmer van IL-1.
Recessief of dominant? De relatie tussen genotype en fenotype is dus complex. De meeste patiënten hebben 2 gemuteerde MEFV-allelen. Daarbij kan de patiënt homozygoot zijn voor 1 mutatie of samengesteld heterozygoot, waarbij er in ieder allel een andere mutatie is. Bij ruim een derde van de patiënten is slechts 1 mutant allel aantoonbaar. Een patiënt is dan heterozygoot en toch ziek, een situatie die past bij autosomaal dominante aandoeningen. Bij een deel van de FMF-patiënten zijn zelfs beide MEFV-allelen niet-afwijkend.6 Omgekeerd wordt niet iedereen die 2 FMF-mutaties heeft ziek. Gezonde familieleden blijken soms precies dezelfde 2 mutaties te hebben als de patiënt.7
De 5 meest voorkomende MEFV-mutaties zijn p.M694I, p.M694V, p.M680I, p.V726A en p.E148Q. Vooral de laatste mutatie is doorgaans onvoldoende om ziekteverschijnselen te veroorzaken. De eerste 4 mutaties, in het bijzonder p.M694V, daarentegen zijn geassocieerd met een ernstiger ziektebeloop. Andere erfelijke factoren, zoals het geslacht, kunnen de ziekte-ernst ook beïnvloeden. Omgevingsfactoren zijn mede bepalend voor de ernst van symptomen en het risico op het optreden van amyloïdose. Zo verloopt de ziekte ernstiger bij patiënten uit landen met een hoge zuigelingensterfte, een maat voor sociaal-economische ontwikkeling.8 Mogelijk speelt de hogere infectiedruk in het land van herkomst een rol doordat ontstekingsreacties frequenter worden uitgelokt. Turken in Turkije hebben inderdaad ernstigere ziekteverschijnselen dan Duitse patiënten van Turkse afkomst.9
Behandeling
Een van de majeure criteria is een goede klinische respons op colchicine, die bij zeker 95% van de patiënten optreedt.4 Onderhoudsbehandeling met colchicine voorkomt de aanvallen effectief bij 65% en vermindert de ernst en frequentie van de aanvallen bij nog eens 30% van de patiënten.10 Uit een grote niet-gecontroleerde prospectieve studie blijkt dat mensen die consequent colchicine gebruiken beschermd zijn tegen het ontwikkelen van amyloïdose. Als tijdens de behandeling de waarden van ontstekingseiwitten als SAA tussen de aanvallen niet verhoogd zijn, wijst dat op effectieve preventie van amyloïdose. Het gebruik van colchicine is veilig gebleken voor pediatrische patiënten, zwangeren en hun ongeboren kind. De behandeling moet in principe levenslang worden voortgezet.
Voor de enkele patiënt die geen baat heeft bij colchicine of daarvan te veel bijwerkingen ondervindt, bestaat geen geregistreerde therapie. Vanwege de centrale rol van IL-1β in de pathogenese is IL-1-blokkade geprobeerd. Uit gepubliceerde casusbeschrijvingen blijkt dat de ziekte bij patiënten die deze offlabel-behandeling kregen in remissie kwam, net als bij patiënt B. Met een onderhoudsbehandeling met anakinra heeft zij in de afgelopen 8 jaar slechts zeer incidentele aanvallen gehad.
Tijdens de follow-up van deze patiënten is het verstandig om ten minste 1 keer per jaar een urineportie te controleren op proteïnurie, ook bij patiënten die ogenschijnlijk niet of nauwelijks meer aanvallen doormaken. Proteïnurie is de vroegste uiting van de ontwikkeling van AA-amyloïdose. Als deze aandoening in een vroeg stadium ontdekt wordt, kan de progressie tot nierfalen vaak worden voorkomen door de anti-inflammatoire behandeling te intensiveren. Het is verstandig de patiënt hiervoor te verwijzen naar een gespecialiseerde kliniek. Bij sommige patiënten is amyloïdose in een vroeg stadium zelfs nog reversibel.
Dames en Heren, het herkennen van familiaire Middellandse Zeekoorts kan in de praktijk moeilijk zijn, zowel bij kinderen als bij volwassenen. Artsen moeten de diagnose overwegen wanneer er perioden van volledig klinisch herstel zijn tussen aanvallen van ziekte. Tijdens deze aanvallen heeft de patiënt bijna altijd koorts en vaak pijn in de buik, thorax of gewrichten, en zijn de ontstekingsparameters verhoogd. Tijdige behandeling met colchicine en zo nodig met interleukine-1-blokkade kan de ziekte bij vrijwel alle patiënten onder controle brengen. Dit geeft patiënten niet alleen een betere kwaliteit van leven, maar behoedt hen op termijn ook voor het ontwikkelen van amyloïdose en terminale nierinsufficiëntie. Als de diagnose te laat wordt gesteld, kan niervervangende behandeling nodig zijn.
Leerpunten
-
Middellandse Zeekoorts is een erfelijke ontstekingsziekte en komt vooral voor bij Turkse en Marokkaanse allochtonen in Nederland.
-
Atypische presentaties, bijvoorbeeld het ontbreken van koorts, kunnen tot een vertraagde diagnose leiden.
-
‘Middellandse Zeekoorts’ is een klinische diagnose. Deze kan worden ondersteund met genetisch onderzoek, maar dit is niet altijd doorslaggevend.
-
Behandeling met colchicine is doorgaans effectief, maar patiënten die niet of niet op tijd worden behandeld kunnen amyloïdose en terminale nierinsufficiëntie ontwikkelen.
-
Wanneer behandeling met colchicine tekortschiet, kan interleukine-1-blokkade uitkomst bieden.
Literatuur
Ben-Chetrit E, Touitou I. Familial Mediterranean fever in the world. Arthritis Rheum. 2009;61:1447-53 Medline. doi:10.1002/art.24458
Samuels J, Aksentijevich I, Torosyan Y, et al. Familial Mediterranean fever at the millennium. clinical spectrum, ancient mutations, and a survey of 100 American referrals to the National Institutes of Health. Medicine (Baltimore). 1998;77:268-97 Medline. doi:10.1097/00005792-199807000-00005
Ben-Zvi I, Livneh A. Chronic inflammation in FMF: markers, risk factors, outcomes and therapy. Nat Rev Rheumatol. 2011;7:105-12 Medline.
Livneh A, Langevitz P, Zemer D, et al. Criteria for the diagnosis of familial Mediterranean fever. Arthritis Rheum. 1997;40:1879-85 Medline. doi:10.1002/art.1780401023
Chae JJ, Cho YH, Lee GS, et al. Gain-of-function pyrin mutations induce NLRP3 protein-independent interleukin-1beta activation and severe autoinflammation in mice. Immunity. 2011;34:755-78 Medline. doi:10.1016/j.immuni.2011.02.020
Özen S. Changing concepts in familial Mediterranean fever: Is it possible to have an autosomal-recessive disease with only one mutation? Arthritis Rheum. 2009;60:1575-7 Medline. doi:10.1002/art.24565
Camus D, Shinar Y, Aamar S, et al. ‘Silent’ carriage of two familial Mediterranean fever gene mutations in large families with only a single identified patient. Clin Genet. 2012;82:288-91 Medline. doi:10.1111/j.1399-0004.2011.01785.x
Touitou I, Sarkisian T, Medlej-Hashim M, et al. Country as the primary risk factor for renal amyloidosis in familial Mediterranean fever. Arthritis Rheum. 2007;56:1706-12 Medline. doi:10.1002/art.22507
Ozen S, Aktay N, Lainka E, Duzova A, Bakkaloglu A, Kallinich T. Disease severity in children and adolescents with familial Mediterranean fever: A comparative study to explore environmental effects on a monogenic disease. Ann Rheum Dis. 2009;68:246-8 Medline. doi:10.1136/ard.2008.092031
Zemer D, Pras M, Sohar E, Modan M, Cabili S, Gafni J. Colchicine in the prevention and treatment of the amyloidosis of familial Mediterranean fever. N Engl J Med. 1986;314:1001-5 Medline. doi:10.1056/NEJM198604173141601
Artikelinformatie
Citeer dit artikel als



Reacties