
Samenvatting
De diagnose ‘vasculitis’ vereist in het algemeen een histopathologische onderbouwing mede doordat andere ziektebeelden sterk op vasculitis kunnen gelijken.
Vasculitis kan primair of idiopathisch zijn, maar ook verband houden met andere ziekten (secundaire vasculitis), voornamelijk met sommige infecties.
De primaire vasculitiden worden ingedeeld op grond van de grootte van de aangedane vaten en de aard van de ontsteking in combinatie met de klinische symptomen.
De ontdekking van autoantistoffen, met name antineutrofielencytoplasma-antistoffen (ANCA), bij een aantal van deze primaire vasculitiden heeft niet alleen de diagnostiek gefaciliteerd, maar heeft tevens tot meer inzicht in de pathofysiologie geleid.
De behandeling van primaire vasculitis bestaat uit toediening van corticosteroïden al of niet in combinatie met immuunsuppressiva.
Nieuwe inzichten in de pathofysiologie kunnen specifiekere en minder toxische behandelingswijzen mogelijk maken.
artikel
Vasculitis kan gedefinieerd worden als een ontsteking van bloedvaten. De klinische manifestatie is uitermate variabel, afhankelijk van de plaats en het kaliber van de aangedane bloedvaten en van de aard en de uitgebreidheid van de ontsteking. Vanwege de variabele en vaak ook atypische presentatie wordt de diagnose niet zelden laat gesteld. In dit artikel beschrijven wij de klinische indeling van de vasculitiden met aandacht voor pathogenetische momenten. Ook gaan wij in op diagnostiek en behandeling. Ter illustratie volgt allereerst een patiëntenbeschrijving.
Een 36-jarige vrouw met een blanco voorgeschiedenis maakte in mei een kaakontsteking door die spontaan herstelde. In september kreeg zij verspringende spier- en gewrichtsklachten. Vanwege persisterende pijn met zwelling van de linker enkel bezocht zij haar huisarts. Bloedonderzoek liet een positieve uitslag voor reumafactoren zien. De diagnose ‘beginnende reumatoïde artritis’ werd overwogen, waarna behandeling volgde met ibuprofen. De klachten bleven bestaan, patiënte kreeg subfebriele temperatuur en haar gewicht nam af met 6 kg zonder aanwijsbare oorzaak. Zij werd verwezen naar ons ziekenhuis voor nader onderzoek. Bij lichamelijk onderzoek werd een matig zieke vrouw gezien, met een rectaal gemeten temperatuur van 38,1°C, een bloeddruk van 135/75 mmHg, een polsfrequentie van 120/min met enkele extrasystolen. De neus was enigszins verstopt en er was enige purulente neusuitvloed. Over het hart werd een holosystolische souffle (graad 2-6) gehoord, maximaal aan de apex. Patiënte had bewegingsbeperking van de linker heup, op de onderbenen waren enkele purpu ra-plekjes aanwezig.
Laboratoriumonderzoek toonde: BSE: 119 mm in het 1e uur; hemoglobineconcentratie: 5,9 mmol/l; leukocytenaantal: 14,7 × 109/l; serumcreatinine: 104 ?mol/l. In het urinesediment waren veel erytrocyten en enkele erytrocytencilinders aanwezig, alsmede 15-20 leukocyten per gezichtsveld. De 24-uursurine bevatte 2,1 g eiwit. De concentratie van C-reactief proteïne bedroeg 135 mg/l, de IgM-reumafactoruitslag was positief (70 kIU/l), een test voor antineutrofielencytoplasma-antistoffen (ANCA) was positief in een titer van 1:160 met een perinucleair fluorescentiepatroon (p-ANCA), cryoglobulinen en antinucleaire antistoffen waren niet aantoonbaar.
Histopathologisch onderzoek van een stansbiopt uit één van de purpura liet leukocytoclastische vasculitis zien (vaatwandbeschadiging met infiltratie met polymorfkernige granulocyten) met immuundeposities in de vaatwandjes. De combinatie van symptomen tezamen met een positieve p-ANCA-uitslag deed systemische vasculitis vermoeden. Drie bloedkweken leverden echter groei van Streptococcus viridans op, terwijl bij transoesofageale echocardiografie een insufficiënte mitraalklep met vegetaties werd gezien.
Met als diagnose ‘subacute bacteriële endocarditis’ werd patiënte gedurende 6 weken intraveneus met antibiotica behandeld. Hierop volgde volledig herstel en werden alle afwijkende laboratoriumwaarden weer normaal.
klinische indeling van vasculitis
Vasculitiden kunnen allereerst ingedeeld worden in de categorieën (a) primaire of idiopathische vasculitiden, (b) vasculitiden in het kader van andere ziekten, (c) ziektebeelden die kunnen lijken op vasculitis (‘vasculitis-look-alike’).
Primaire vasculitiden
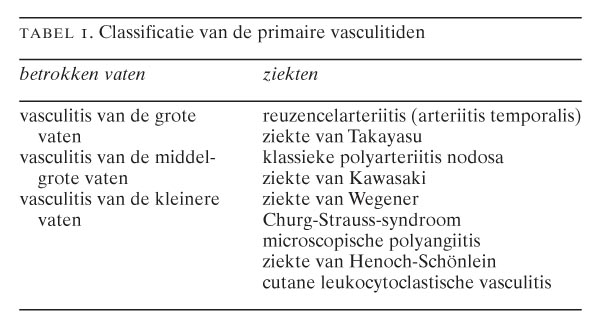
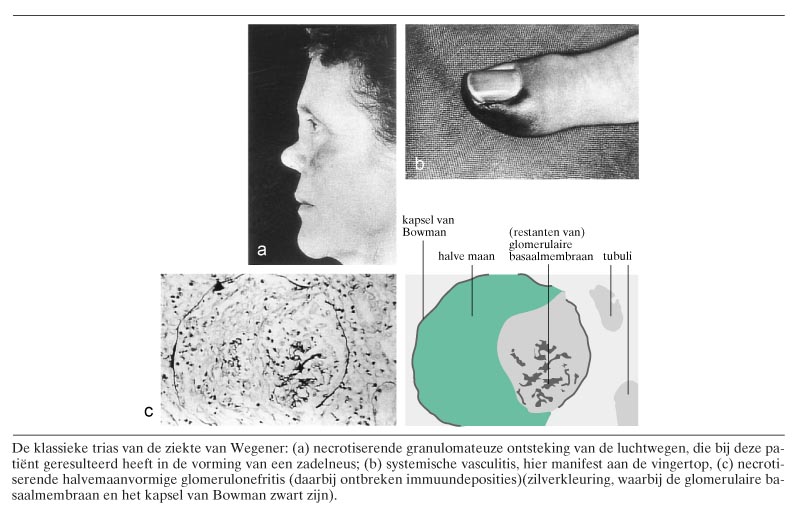
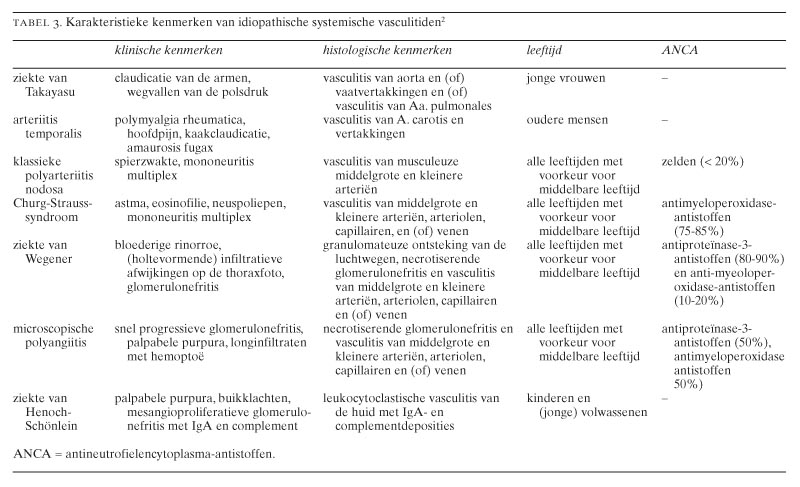
De primaire vasculitiden worden ingedeeld op grond van de grootte van de aangedane vaten, de aard van de ontsteking en de klinische symptomen (tabel 1). Door de American College of Rheumatologists (ACR) zijn classificatiecriteria opgesteld voor de primaire vasculitiden.1 Deze criteria zijn bedoeld om patiënten met bewezen vasculitis, dat wil zeggen histopathologisch of (bij uitzondering) angiografisch aangetoonde vasculitis, verder te classificeren naar omschreven ziekte-entiteiten. Deze criteria kunnen niet gebruikt worden voor diagnostische doeleinden, wat duidelijk wordt wanneer wij bijvoorbeeld de criteria voor de classificatie van de ziekte van Wegener bezien (tabel 2): iedere patiënt met microscopische hematurie en purulente of bloederige neusuitvloed zou, indien de criteria diagnostisch gebruikt worden, de diagnose ‘ziekte van Wegener’ krijgen (figuur 1). Zelfs wanneer vasculitis histopathologisch is aangetoond, zoals bij de aan het begin beschreven casus, zouden patiënten ten onrechte die diagnose krijgen. Onze patiënt vertoonde microscopische hematurie, purulente neusuitvloed en leukocytoclastische vasculitis van de huid, maar bleek later toch een subacute bacteriële endocarditis te hebben. Vanwege deze tekortkomingen van de ACR-criteria zijn onlangs definities opgesteld voor de verschillende vasculitiden.2 Deze definities zijn vooral gebaseerd op de histopathologische bevindingen in de afwijkingen en op de grootte van de aangedane vaten. In de klinische praktijk blijken deze aldus gedefinieerde ziektebeelden niet altijd eenduidig aanwijsbaar. Mengbeelden van de genoemde vasculitiden komen veelvuldig voor. Met inachtneming van deze beperkingen geeft tabel 3 de karakteristieken weer van de idiopathische vasculitiden.
Vasculitiden in het kader van andere ziekten
De casus waarmee dit overzicht begon, geeft een voorbeeld van secundaire vasculitis: bacteriële endocarditis leidde tot systemische vasculitis met als klinische uitingsvormen purpura, artritis, en glomerulonefritis. Bij de pathofysiologie spelen verschillende factoren een rol: bacteriële emboli kunnen afsluiting van kleine arteriën en arteriolen geven met infarcering; voorts kan depositie van immuuncomplexen vanuit de circulatie of in-situvorming van immuuncomplexen, veelal bestaande uit bacteriële antigenen en daartegen gerichte antistoffen, leiden tot vaatwandontsteking in de microcirculatie van de subcutis (purpura) en in de glomerulaire vaatkluwen; tenslotte kunnen reumafactoren, al of niet in de vorm van cryoglobulinen, bijdragen aan immuuncomplexvorming. Deze reumafactoren ontstaan als gevolg van de chronische immuunstimulatie waarmee de bacteriële endocarditis gepaard gaat.
Vele vormen van secundaire vasculitis zijn inderdaad op te vatten als immuuncomplexziekten. Daarbij spelen microbiële antigenen een belangrijke rol. Ook niet-microbiële antigenen kunnen aan immuuncomplexvorming ten grondslag liggen, zoals in het klassieke beeld van serumziekte: daarbij leiden heterologe eiwitten, bijvoorbeeld in paardenserum, na injectie tot hiertegen gerichte antistoffen, die weer met deze heterologe eiwitten kunnen complexeren. Het therapeutisch gebruik van polyklonale (en monoklonale) antistoffen kan zo tot verschijnselen van serumziekte leiden.
Ook autoantigenen kunnen bij immuuncomplexvorming betrokken zijn. Vasculitis en glomerulonefritis bij lupus erythematodes disseminatus of bij reumatoïde artritis berusten eveneens op immuuncomplexvorming waarbij respectievelijk kernbestanddelen (DNA, nucleohistonen)3 en immuunglobulinen zelf (waartegen reumafactoren gericht zijn) als antigeen kunnen fungeren. Behalve secundaire vormen van vasculitis, die berusten op immuuncomplexvorming, zijn er ook vormen van vasculitis die berusten op directe invasie van de vaatwand door micro-organismen.4 Bespreking van deze situaties, waarbij onder andere mycotische aneurysmata kunnen ontstaan, valt buiten het bestek van dit artikel.
Op vasculitis lijkende ziektebeelden
Een aantal klinische entiteiten kan zich manifesteren als vasculitis, maar berust niet op vaatwandontsteking in strikte zin. Voorbeelden hiervan zijn cholesterolembolieën en het antifosfolipidesyndroom,5 waarbij trombosering in arteriën en venen het opvallendst zijn. Herkenning van deze entiteiten is belangrijk, niet alleen om redenen van pathofysiologisch inzicht, maar vooral vanwege behandeling en prognosestelling.
diagnose
Histopathologisch onderzoek
Uit het voorgaande moge duidelijk zijn dat histopathologisch onderzoek de hoeksteen is bij de diagnostiek van vasculitis. Voor het verkrijgen van geschikt weefsel kan de volgende benadering gevolgd worden:
biopteren van zichtbare afwijkingen, zoals purpura van de huid en afwijkingen van de slijmvliezen in de bovenste luchtwegen;
biopteren van organen die bij beeldvormend onderzoek, functietests of laboratoriumonderzoek afwijkingen tonen, bijvoorbeeld een nierbiopt bij urinesedimentafwijkingen compatibel met glomerulonefritis en een zenuwbiopt bij bevindingen passend bij mononeuritis multiplex.
in enkele gevallen kan een zogenaamd ‘blind’ biopt worden genomen, dat meestal een huid-spier-fasciebiopt betreft; de opbrengst van een dergelijk biopt bij de diagnostiek van vasculitis is echter laag. Een uitzondering hierop vormt de biopsie van de A. temporalis bij klinisch vermoeden van arteriitis temporalis ook zonder dat palpabele afwijkingen, dat wil zeggen verdikking of het ontbreken van pulsaties, aanwezig zijn.
Soms beeldvormend onderzoek
In twee gevallen kan beeldvormend onderzoek het histopathologisch onderzoek vervangen. Het eerste betreft de ziekte van Takayasu waarbij angiografie van de aortaboog met zijn vertakkingen een typisch beeld van vernauwingen met soms aneurysmatische verwijdingen laat zien; het tweede geval is de klassieke polyarteriitis nodosa indien geen histopathologische diagnose verkregen kan worden. Angiografie van de viscerale vaten en (of) nierarteriën kan typische microaneurysmata laten zien; deze zijn echter niet pathognomonisch voor dit ziektebeeld, aangezien microaneurysmata ook kunnen voorkomen bij andere vormen van vasculitis zoals de ziekte van Wegener, het Churg-Strauss-syndroom en microscopische polyangiitis.
Wanneer de diagnose ‘vasculitis’ op histopathologische gronden, of bij uitzondering door middel van angiografie, is gesteld, dient classificatie plaats te vinden. De mogelijkheid van secundaire vormen (zie eerder) dient altijd in overweging genomen te worden, in het bijzonder moet men daarbij denken aan vasculitis in relatie tot infectieziekten (zoals in onze casus) en aan vasculitis in het kader van een systemische auto-immuunziekte. Bij leukocytoclastische vasculitis die zich beperkt tot de huid dient ook altijd aan overgevoeligheid voor geneesmiddelen gedacht te worden. Voor de classificatie van de primaire vasculitiden dient men niet alleen het genoemde histopathologisch onderzoek te doen, maar moet men ook speciaal letten op kenmerkende symptomen zoals beschreven in tabel 3. De klinische manifestatie is echter vaak atypisch en kan leiden tot vertraging in de diagnosestelling.6 Voor de classificatie van de primaire vasculitiden kan gebruikgemaakt worden van de ACR-criteria,1 mits eerst het histopathologische bewijs van vasculitis geleverd is.
Antineutrofielencytoplasma-antistoffen
Een bijzondere plaats bij de diagnostiek van primaire vasculitiden is weggelegd voor de bepaling van de ANCA.7 Bij de ziekte van Wegener werden als eerste autoantistoffen aangetoond, die bij indirecte immuunfluorescentie een typisch cytoplasmatisch aankleuringspatroon laten zien op met ethanol gefixeerde granulocyten (c-ANCA).8 Deze c-ANCA bleken gericht te zijn tegen proteïnase 3, een serineprotease uit de azurofiele korrels van myeloïde cellen. Sera van sommige patiënten met de ziekte van Wegener lieten bij indirecte immuunfluorescentie een perinucleaire aankleuring van de granulocyt zien (p-ANCA).8 De p-ANCA bij patiënten met systemische vasculitis bleken in de meeste gevallen gericht tegen myeloperoxidase en in enkele gevallen tegen elastase, beide enzymen eveneens afkomstig uit de azurofiele korrels. Terwijl anti-proteïnase-3-antistoffen vooral voorkomen bij patiënten met de ziekte van Wegener, komen antimyeloperoxidaseantistoffen voor bij verschillende vormen van systemische vasculitis (zie tabel 3). Beide hebben echter een hoge specificiteit voor de primaire vasculitiden. De p-ANCA met andere antigeenspecificiteit, zoals tegen lactoferrine en tegen andere nog slechts ten dele gekarakteriseerde bestanddelen van myeloïde cellen, blijken echter voor te komen bij andere idiopathische ontstekingsziekten, zoals reumatoïde artritis en inflammatoire darmziekten, en tevens bij sommige chronisch verlopende infecties, zoals onze casus illustreert.7 8 De diagnostische betekenis van p-ANCA als zodanig is dus beperkt. Dit betekent dat een positieve ANCA-test altijd gevolgd dient te worden door specifieke tests voor antiproteïnase-3- en antimyeloperoxidaseantistoffen. Vooral bij patiënten met de ziekte van Wegener is gebleken dat stijging van de hoeveelheid ANCA in het bloed vaak gevolgd wordt door een exacerbatie van de ziekte. Dit geldt in het bijzonder wanneer een stijging optreedt van de IgG3-subklasse van deze autoantistoffen. Of stijging van ANCA-spiegels alleen voldoende reden is om behandeling in te stellen is nog omstreden.9 Een dergelijke stijging dient de clinicus echter wel alert te maken op de mogelijkheid van een naderende exacerbatie.
Ernst van de ziekte
Wanneer eenmaal de diagnose ‘vasculitis’ is gesteld en de ziekte geclassificeerd is, dienen de uitgebreidheid en de activiteit van de ziekte te worden vastgesteld. De eventuele betrokkenheid van orgaansystemen dient vastgesteld te worden door middel van biochemisch, functioneel en afbeeldend onderzoek. Criteria voor ernstige (‘major’) en minder ernstige (‘minor’) exacerbaties zijn opgesteld teneinde bepaalde behandelingswijzen (zie verder) te rechtvaardigen.10 Voorts zijn ziekteactiviteitsscores ontwikkeld om de ernst van de ziekte en de respons op behandeling te kunnen kwantificeren.11
pathogenese
Zoals eerder genoemd, is de pathogenese van de secundaire vasculitiden in veel gevallen, althans ten dele, opgehelderd. Immuuncomplexen, neergeslagen vanuit de circulatie of in situ gevormd, spelen hier een belangrijke rol. Bij de primaire vasculitiden, met uitzondering van die gevallen van klassieke polyarteritis nodosa en gemengde cryoglobulinemie verband houdende met hepatitis-B- en -C-virusinfectie, is de pathogenese nog grotendeels onopgehelderd. Immuundeposities zijn veelal niet aanwezig. De ontdekking van ANCA heeft echter een enorme stimulans gegeven aan de ontrafeling van de pathofysiologie. ANCA blijken in vitro in staat granulocyten te activeren en de interactie tussen ontstekingscellen en endotheel te vergemakkelijken.12 Hiervoor moeten granulocyten wel eerst gepreactiveerd zijn door voorbehandeling met kleine hoeveelheden van bijvoorbeeld tumornecrosisfactor-?, een pro-inflammatoir cytokine. Deze preactivering of ‘priming’ leidt tot expressie van de doelwitantigenen van ANCA op de celmembraan, waarna de autoantistof hieraan bindt en de granulocyt verder activeert, wat leidt tot uitstoten van lysosomale enzymen en zuurstofradicalen en tot lysis van eventueel aanwezige endotheelcellen. Dierexperimenteel onderzoek heeft laten zien dat bij de rat perfusie van de nier met de producten van geactiveerde granulocyten bij aanwezigheid van antimyeloperoxidaseantistoffen leidt tot necrotiserende glomerulonefritis met glomerulaire zogenaamde cellige halve manen; bij systemische perfusie ontstaat vasculitis in darm en longen.13 De aanwezigheid van ANCA alleen is echter onvoldoende om het ziekteproces te induceren, zowel in het proefdier als bij de mens. Een externe factor die tot priming van ontstekingscellen leidt, lijkt hiervoor tevens nodig. In dit verband is van belang dat exacerbaties van vasculitis, in het bijzonder de ziekte van Wegener, nogal eens voorafgegaan worden door infecties. In een recent onderzoek vonden wij dat chronisch dragerschap van Staphylococcus aureus een belangrijke onafhankelijke risicofactor vormt voor het optreden van exacerbaties bij de ziekte van Wegener.14 De waarneming dat preventieve onderhoudsbehandeling met co-trimoxazol (960 mg 2 dd) leidt tot 68 reductie van recidieven van de ziekte van Wegener doet ook vermoeden dat microbiële factoren, zoals dragerschap van S. aureus, een rol spelen bij de expressie van de ziekte.15
behandeling
Behandeling van secundaire vormen van vasculitis dient gericht te zijn op de onderliggende aandoening (zoals bij de casus bij het begin van dit artikel) en blijft hier buiten beschouwing. Aangezien de oorzaak van de primaire vasculitiden niet bekend is, zal de behandeling hierbij vooral gericht zijn op het niet-specifiek onderdrukken van de ontstekingsreactie. Bij wijze van voorbeeld zal de behandeling van de ziekte van Wegener worden besproken. De gemiddelde overlevingsduur van patiënten met deze ziekte bedroeg slechts 5 maanden voordat corticosteroïden en immuunsuppressiva beschikbaar waren. Corticosteroïden alleen gaven een verlenging tot 12 maanden. De combinatie van cyclofosfamide 2-3 mg/kg per dag met prednison 1 mg/kg per dag, beide in orale vorm, is een belangrijke aanwinst gebleken. Het merendeel der patiënten blijkt hiermee in remissie te komen. Plasmaferese en (of) stootdoses methylprednisolon 1 g i.v. gedurende 3 dagen, kunnen aangewezen zijn bij zeer ernstige manifestatie van de ziekte. Hoewel bij de meeste patiënten aldus remissie van de ziekte bereikt kan worden, treedt bij circa 50 bij follow-up opflikkering van de ziekte op. Om deze reden wordt aanbevolen de behandeling met cyclofosfamide langdurig, dat wil zeggen minimaal één jaar, te continueren. Nadeel hiervan is de grote kans op opportunistische infecties en in mindere mate op maligniteiten. Het zou daarom van groot belang zijn wanneer tevoren bepaald zou kunnen worden welke patiënten een grote kans op opflikkering hebben. Gebleken is, zoals wij al schreven, dat in het bijzonder patiënten die drager zijn van S. aureus een sterk verhoogde kans op opflikkering hebben en dat onderhoudsbehandeling met co-trimoxazol de kans op recidief belangrijk doet afnemen.14 Voorts is het persisteren van ANCA ten tijde van remissie een risicofactor voor recidief van de ziekte, terwijl stijging van de titer van ANCA het optreden ervan kan aankondigen. Vroegtijdige behandeling, aan te vangen ten tijde van ANCA-stijging, blijkt het recidief in veel gevallen te kunnen voorkomen.
Uit het voorgaande is duidelijk dat verdieping van inzicht in de pathogenese van de systemische vasculitiden kan leiden tot rationelere en specifieke behandelingswijzen die met minder toxiciteit gepaard gaan. In Europees verband wordt thans een aantal van deze therapieën getest op hun klinische bruikbaarheid. De uitkomsten hiervan zullen het ons hopelijk mogelijk maken sterfte en morbiditeit door deze groep van ziekten te verminderen.
Literatuur
Hunder GG, Arend WP, Bloch DA, Calabrese LH, Fauci AS,Fries JF, et al. The American College of Rheumatology 1990 criteria for theclassification of vasculitis. Introduction. Arthritis Rheum 1990;33:1065-7.
Jennette JC, Falk RJ, Andrassy K, Bacon PA, Churg J, GrossWL, et al. Nomenclature of systemic vasculitides. Proposal of aninternational consensus conference. Arthritis Rheum 1994;37:187-92.
Berden JM. Immunologie in de medische praktijk. III. Lupuserythematodes disseminatus: gestoorde apoptose?Ned Tijdschr Geneeskd1997;141:1848-54.
Somer T, Finegold SM. Vasculitides associated withinfections, immunization, and antimicrobial drugs. Clin Infect Dis1995;20:1010-36.
Hughes GRV. The antiphospholipid syndrome: ten years on.Lancet 1993;342:341-4.
Cohen Tervaert JW, Woude FJ van der, Kallenberg CGM.Analyse van de symptomen voorafgaand aan de diagnose ziekte van Wegener.Ned Tijdschr Geneeskd1987;131:1391-4.
Kallenberg CGM, Brouwer E, Weening JJ, Cohen Tervaert JW.Anti-neutrophil cytoplasmic antibodies: current diagnostic andpathophysiological potential. Kidney Int 1994;46:1-15.
Franssen CFM, Hoorntje SJ, Kallenberg CGM, Gans ROB. Deplaats van perinucleaire antistoffen tegen het cytoplasma van neutrofielegranulocyten (p-ANCA) bij de diagnostiek van vasculitis en chronischeinflammatoire ziekten. Ned TijdschrGeneeskd 1996;140:2076-82.
Cohen Tervaert JW, Huitema MG, Hené RJ, Sluiter WJ,The TH, Hem GK van der, et al. Prevention of relapses in Wegener'sgranulomatosis by treatment based on antineutrophil cytoplasmic antibodytitre. Lancet 1990;336:709-11.
Kallenberg CGM, Cohen Tervaert JW, Stegeman CA. Criteriafor disease activity in Wegener's granulomatosis: a requirement forlongitudinal clinical studies. APMIS 1990;98(Suppl 19):37-9.
Luqmani RA, Bacon PA, Moots RJ, Janssen BA, Pall A, EmeryP, et al. Birmingham Vasculitis Activity Score (BVAS) in systemic necrotizingvasculitis. QJM 1994;87:671-8.
Kallenberg CGM, Brouwer E, Mulder AHL, Stegeman CA,Weening JJ, Cohen Tervaert JW. ANCA pathophysiology revisited. ClinExp Immunol 1995;100:1-3.
Heeringa P, Brouwer E, Cohen Tervaert JW, Weening JJ,Kallenberg CGM. Animal models of anti-neutrophil cytoplasmic antibodyassociated vasculitis. Kidney Int ter perse.
Stegeman CA, Cohen Tervaert JW, Sluiter WJ, Manson WL,Jong PE de, Kallenberg CGM. Association of chronic nasal carriage ofStaphylococcus aureus and higher relapse rates in Wegener'sgranulomatosis. Ann Intern Med 1994;120:12-7.
Stegeman CA, Cohen Tervaert JW, Jong PE de, KallenbergCGM. Trimethoprim-sulfamethoxazole (co-trimoxazole) for the prevention ofrelapses of Wegener's granulomatosis. Dutch Co-Trimoxazole Wegener StudyGroup. N Engl J Med 1996;335:16-20.
Artikelinformatie
Citeer dit artikel als




{kind=link}
{kind=link}
{kind=link}
{kind=link}
Reacties