
Samenvatting
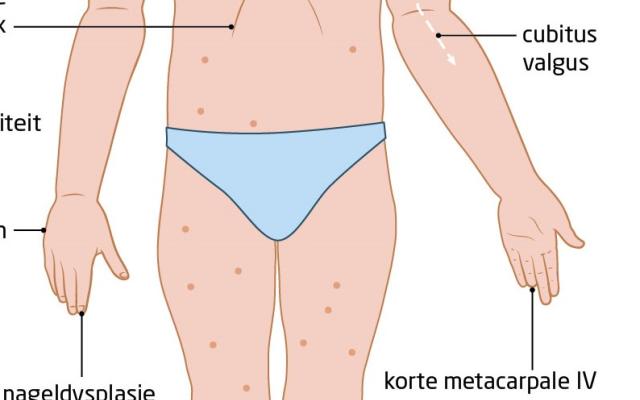
- Het Noonan-syndroom is een relatief frequent voorkomende autosomaal dominante aandoening met als belangrijkste problemen: hartafwijkingen, een kleine lengte, voedingsproblemen in het 1e jaar en later leer- en gedragsproblemen.
- De diagnose is klinisch, al kan in 50 van de gevallen de diagnose bevestigd worden met een mutatie in het proteïnetyrosinefosfatase(PTPN11)-gen.
- Studies naar het effect van groeihormoonbehandeling op de eindlengte laten nog geen definitieve conclusies toe. Deze behandeling dient vooralsnog in studieverband plaats te vinden.
- Vroegkinderlijke voedingsproblemen zijn lastig te hanteren, doch verdwijnen spontaan en lijken de groei niet negatief te beïnvloeden.
- Gedrags- en leerproblemen met de kenmerken van een non-verbale leerstoornis komen frequent voor en vragen om een specifieke aanpak.
Artikelinformatie
Citeer dit artikel als



Reacties