
artikel
Inleiding
de ziekte
Myotone dystrofie type 1, of de ziekte van Steinert, behoort tot de erfelijke spierdystrofieën, maar vele organen, zoals het hart, de tractus digestivus, de zintuigen en het centrale zenuwstelsel kunnen eveneens aangedaan zijn.1 Klinisch worden er 4 ziektetypen onderscheiden: het ‘milde’ type, het volwassen type, het kindertype en het congenitale (aangeboren) type (tabel).
Het volwassen type, met een beginleeftijd tussen 12 en 50 jaar, veroorzaakt het klassieke ziektebeeld met als kenmerken myotonie (het niet snel kunnen ontspannen van de spieren na aanspannen), een langzaam progressieve spierzwakte en orgaancomplicaties.1 De spierzwakte begint in het gelaat en de distale spieren van de extremiteiten en breidt zich in de loop van 20-30 jaar uit tot de proximale arm- en beenspieren (figuur 1). Bijna de helft van de patiënten wordt in de laatste jaren voor hun dood rolstoelafhankelijk.2 Het geleidingssysteem van het hart is aangedaan bij ruim 80 van de patiënten, resulterend in een ritme- of geleidingsstoornis, en acute hartdood is frequent.2 Slikproblemen, een vertraagde maagontlediging, buikpijn, obstipatie en diarree, veroorzaken veel last en zijn niet makkelijk behandelbaar. De mentale stoornissen, die een gevolg zijn van de aantasting van de hersenen, bestaan bij het volwassen type uit apathie, verlies van initiatief en een toegenomen slaapzucht. Berucht is de grotere kans op complicaties die kunnen optreden na algehele anesthesie en tijdens de zwangerschap.
Bij de kindervorm zijn leer- en gedragsproblemen aanvankelijk het enige symptoom. Het congenitale type wordt gekenmerkt door een ernstige spierzwakte die al prenataal aanwezig is. Ademhalings- en slikproblemen, die post partum levensbedreigend kunnen zijn, verminderen in de loop van weken of maanden. Vervolgens zijn de motorische en de verstandelijke ontwikkeling vertraagd.3
Myotone dystrofie type 1 is autosomaal dominant erfelijk, maar er treedt een aantal bijzondere verschijnselen op bij de overerving, zoals anticipatie (dat wil zeggen een vroegere beginleeftijd gepaard gaande met een ernstiger beloop in opeenvolgende generaties) en een vrijwel exclusieve overerving via de moeder van de congenitale vorm.4 Deze bijzondere overerving en de variabiliteit in de klinische presentatie kunnen deels verklaard worden vanuit de moleculaire mechanismen.
De ziekte moet onderscheiden worden van myotone dystrofie type 2, ook ‘proximale myotone myopathie’ genoemd; qua symptomen komen beide ziekten enigszins met elkaar overeen.5
het gen
Myotone dystrofie type 1 behoort net als het fragiele-X-syndroom, de ziekte van Huntington en een aantal vormen van spinocerebellaire ataxie tot de trinucleotide-repeat-ziekten.6 Bij myotone dystrofie type 1 is het aantal herhalingen van een cytosine-thymine-guanine(CTG)-triplet in het gen voor myotone-dystrofieproteïnekinase (DMPK), toegenomen.7 Bij de overerving zijn deze CTG-tripletten instabiel en ze worden meestal langer. In de normale populatie varieert het aantal CTG-tripletten van 5 tot 20. Premutaties, van 35-50 CTG-herhalingen, zijn soms instabiel. Bij tripletlengten van 50-100 spreken wij van een protomutatie, bij meer dan 100 CTG’s van een volledige mutatie. Er is een omgekeerde correlatie tussen het aantal tripletten en de beginleeftijd van de ziekte (zie de tabel).8 De toename van het aantal CTG-herhalingen in een volgende generatie verklaart de ernstiger uiting bij nakomelingen van een aangedane ouder.9 De CTG-tripletten zijn niet alleen instabiel bij de overerving, maar ook op weefselniveau, resulterend in een somatisch mozaïcisme.
Het DMPK-gen bevindt zich op de lange arm van chromosoom 19 op positie q13.3 en is gelegen in een genrijke regio. Het gen bestaat uit 15 exonen en heeft een lengte van 13 kilobasen (kb). De CTG-tripletten bevinden zich in het niet-coderende 3'-deel van het DMPK-gen.7
de eiwitten en de cellen
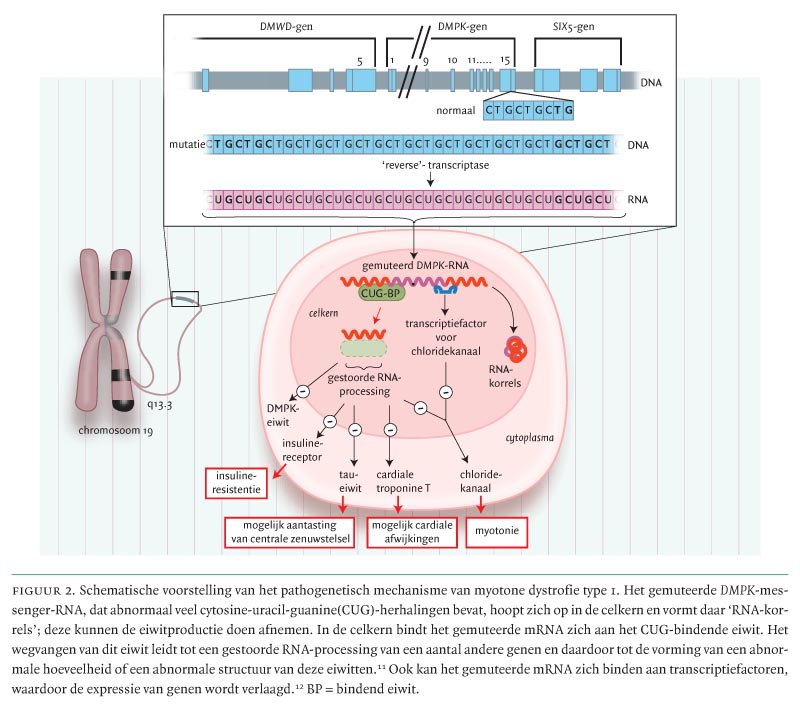
Het pathogenetisch mechanisme van myotone dystrofie type 1 is nog niet opgehelderd, maar het is de laatste jaren duidelijk geworden dat niet een disfunctie van het DMPK-gen zelf, maar vooral het gemuteerde DMPK-messenger-RNA (mRNA) een sleutelrol speelt (figuur 2).10 Het gemuteerde mRNA, dat abnormaal veel cytosine-uracil-guanine(CUG)-herhalingen bevat, hoopt zich op in de celkern en vormt daar ‘RNA-korrels’.11 In de celkern bindt het gemuteerde mRNA zich aan het CUG-bindende eiwit. Het wegvangen van dit eiwit leidt tot een gestoorde RNA-processing van een aantal andere genen, zoals het insulinereceptorgen, het cardiale troponine-T-gen, het spierchloridekanaalgen en het Tau-gen en daardoor tot de vorming van een abnormale hoeveelheid of een abnormale structuur van deze eiwitten.11 Het gemuteerde mRNA kan zich ook binden aan transcriptiefactoren, waardoor de expressie van genen wordt verlaagd. Eén van deze genen codeert voor de chloridekanalen in skeletspieren, die mogelijk betrokken zijn bij de myotonie. Bovengenoemd model kan zowel de autosomaal dominante overerving als veel van de multisysteemkenmerken van de ziekte verklaren.12 Daarnaast is er een verminderde hoeveelheid van het DMPK-eiwit, dat vooral voorkomt in skelet- en hartweefsel en in glad spierweefsel, gevonden.11 De buiten het coderende deel van het gen gelegen verlengde CTG-tripletten leiden niet direct tot een verminderde productie van mRNA. Echter, de ophoping van abnormaal mRNA in de celkern (zie boven) kan wel tot gevolg hebben dat de hoeveelheid mRNA in het cytoplasma afneemt, leidend tot een afname van de eiwitproductie.
Een laatste pathogenetisch mechanisme dat bij myotone dystrofie type 1 wordt verondersteld, is dat de verlengde CTG-tripletten regionaal invloed uitoefenen op de expressie van een aantal genen, waarschijnlijk door verandering van de chromatinestructuur. Verminderde expressie van het aan de 3'-kant van DMPK-gen gelegen SIX5-gen, dat voorkomt in het oog, de skeletspier, het hart en de hersenen, leidt bij muizen tot cataract en hartritmestoornissen. Het DMWD-gen, aan de 5'-kant van het DMPK-gen, komt verminderd tot expressie in cellijnen van patiënten met myotone dystrofie type 1 en heeft invloed op cellulaire processen zoals de celdeling, transcriptie, RNA-processing en signaaltransductie.11
de populatie
Myotone dystrofie type 1 is de meest voorkomende erfelijke neuromusculaire aandoening bij volwassenen. De prevalentie varieert van ongeveer 1 op 8000 in West-Europa en Noord-Amerika tot 1 op 20.000 in Japan. In sommige streken, zoals in een deel van Quebec in Canada, komt de aandoening opvallend veel voor, terwijl de ziekte vrijwel niet voorkomt bij Afrikanen ten zuiden van de Sahara. Mogelijk zijn alle myotone-dystrofie-type-1-mutaties afkomstig van één of enkele ‘founder’-mutatie(s), die in de loop van vele generaties een geleidelijke verlenging hebben ondergaan, tot er een kritische grens werd overschreden waarna ze instabiel werden.3
diagnostiek
De diagnose ‘myotone dystrofie’ is makkelijk te stellen bij een patiënt met klassieke verschijnselen, zoals myotonie, gelaatsspierzwakte en distale spierzwakte. Echter, wanneer de ziekte zich atypisch presenteert, bijvoorbeeld met hartritmestoornissen, buikklachten of leerproblemen bij kinderen, is het lastig, zeker als de ziekte niet eerder in de familie is voorgekomen. De diagnose kan definitief bevestigd worden door het aantonen van de verlengde CTG-tripletten in het DMPK-gen. DNA-onderzoek kan verricht worden in de DNA-diagnostieklaboratoria van de afdelingen Klinische Genetica in de academische ziekenhuizen te Maastricht, Nijmegen en Utrecht. Dit onderzoek heeft een hoge sensitiviteit en specificiteit. DNA-diagnostiek biedt ook duidelijkheid aan familieleden die een verhoogd risico op de aandoening hebben en kan een einde maken aan de onzekerheid of zij wel of geen drager van het gemuteerde gen zijn. Prenatale DNA-diagnostiek is mogelijk, bij voorkeur op vlokken, verkregen via een vlokkentest in de 11e-14e week van de zwangerschap. Als een ernstige uiting van de ziekte te verwachten is, kunnen de aanstaande ouders besluiten tot een abortus provocatus.3
Belangenconflict: geen gemeld. Financiële ondersteuning: geen gemeld.
Literatuur
Brunner HG, Höweler CJ, Smeets HJM, Wieringa B. Een instabiele mutatie als oorzaak van myotone dystrofie. Ned Tijdschr Geneeskd 1993;137:2468-72.
Die-Smulders CEM de, Höweler CJ, Thijs C, Mirandolle JF, Anten HB, Smeets HJM, et al. Age and causes of death in adult-onset myotonic dystrophy. Brain 1998;121(Pt 8):1557-63.
Jennekens FGI, Die-Smulders CEM de, Busch HFM, Höweler CJ. Myotone dystrofie. Begeleiding en behandeling. Maarssen: Elsevier Gezondheidszorg; 2000.
Höweler CJ, Busch HFM, Geraedts JPM, Niermeijer MF, Staal A. Anticipation in myotonic dystrophy: fact or fiction? Brain 1989;112(Pt 3):779-97.
Tieleman AA, Velden MPH van der, Visser MC, Wokke JHJ, Scheffer H, Engelen BGM van. Vier familieleden met proximale myotone myopathie. Ned Tijdschr Geneeskd 2004;148:948-52.
Kremer HPH, Knoers NVAM. Neurodegeneratieve aandoeningen en de rol van trinucleotide-repeat-expansie. I. De ziekten. Ned Tijdschr Geneeskd 1996;140:2325-9.
Groenen P, Wieringa B. Expanding complexity in myotonic dystrophy. Bioessays 1998;20:901-12.
Harley HG, Rundle SA, MacMillan JC, Myring J, Brook JD, Crow S, et al. Size of the unstable CTG repeat sequence in relation to the phenotype and parental transmission in myotonic dystrophy. Am J Hum Genet 1993;52:1164-74.
Harper PS, Harley HG, Reardon W, Shaw DJ. Anticipation in myotonic dystrophy: new light on an old problem. Am J Hum Genet 1992;51:10-6.
Nagamitsu S, Ashizawa T. Myotonic dystrophies. Adv Neurol 2002;88:293-314.
Ranum LPW, Day JW. Myotonic dystrophy: RNA pathogenesis comes into focus. Am J Hum Genet 2004;74:793-804.
Ebralidze A, Wang Y, Petkova V, Ebralidse K, Junghans RP. RNA leaching of transcription factors disrupts transcription in myotonic dystrophy. Science 2004;303:383-7.
Artikelinformatie
Citeer dit artikel als





{kind=link}
{kind=link}
{kind=link}
Reacties