
artikel
Zie ook het artikel op bl. 1556.
Inleiding
Tubereuze sclerose (TS) is een autosomaal dominant overerfelijke neurocutane ziekte met vaak ernstige verschijnselen. Van deze ziekte (met een prevalentie van 1:10.000) is DNA-diagnostiek nog niet mogelijk.1 De eerste beschrijving van een patiënt met dit ziektebeeld betreft een obductieverslag door Von Recklinghausen uit 1862.2 Bourneville beschreef in 1880 voor het eerst gedetailleerd de neurologische symptomen en de macroscopie van de hersenen. Vanwege het knobbelige aspect van de hersenen noemde hij de ziekte TS.3 Vogt beschreef in 1908 een patiënt met epilepsie, mentale retardatie en adenoma sebaceum, een trias die lange tijd als karakteristiek zou worden beschouwd.4 Uit recente studies blijkt, dat deze klassieke trias slechts bij 29 van de patiënten aanwezig is en bij een klein deel zelfs volledig ontbreekt. Door de zeer variabele klachten en verschijnselen kan TS moeilijk te diagnostiseren zijn. Om deze reden heeft Gomez diagnostische criteria opgesteld.5 In de tabellen 1 en 2 worden de verschijnselen aldus onderverdeeld in pathognomonische kenmerken en in verschijnselen die de diagnose pas waarschijnlijk maken als er verschillende kenmerken bij de patiënt te zamen aanwezig zijn.
Genetische aspecten
De overerving van TS is autosomaal dominant met variabele expressie. Elk kind van een TS-patiënt heeft een kans van 50 de aandoening te krijgen. In ongeveer 60 van de gevallen blijken de ouders van een TS-patiënt geen van beiden verschijnselen van de aandoening te hebben.6 De meest waarschijnlijke oorzaak voor het optreden van TS bij het kind is dan een somatische mutatie bij één der ouders, of een mutatie in één der gameten waaruit hun kind met TS is ontstaan. Het herhalingsrisico voor een volgend kind van gezonde ouders van een TS-patiënt wordt gesteld op 1.7
De precieze lokalisatie van het gen voor TS is nog niet bekend. In één onderzoek werden aanwijzingen voor een lokalisatie op chromosoom 9 gevonden, in een ander onderzoek kon dit niet bevestigd worden, maar wel werd een aanwijzing gevonden voor lokalisatie op chromosoom II.8-12 Vroege diagnostiek met behulp van DNA-onderzoek is derhalve nog niet op betrouwbare wijze mogelijk.
Als het gen-locus gevonden is, wordt betrouwbare prenatale diagnostiek mogelijk in gevallen waarbij TS bij meerdere personen in de familie voorkomt. Het is niet te verwachten, dat op zeer korte termijn het gen-defect zelf aantoonbaar zal zijn.
Het is belangrijk de ouders en eventueel overige familieleden voor te lichten over het herhalingsrisico en de variabele expressie van de aandoening.
Dermatologische afwijkingen
Bij patiënten bij wie anamnestisch vermoeden bestaat van TS, levert onderzoek van de huid vaak de eerste aanwijzingen voor de diagnose. Bij 80-90 van de patiënten ziet men witte vlekken, die overigens niet specifiek zijn. Deze hypomelanotische maculae zijn vaak bij de geboorte aanwezig of ontstaan in de eerste levensweken.1314 Ook kunnen deze maculae pas later ontstaan, mogelijk tot zelfs na enige jaren.14 Het aantal maculae varieert van 1 tot 100. De vlekjes kunnen typisch van vorm zijn, zoals die van een esseblad, of zoals de afdruk van een duim, of ‘é confetti’ (klein druppelvormig), maar zijn dit lang niet altijd. De grootte is meestal 1-3 cm. In de eerste levensjaren zijn dit vaak de enige huidafwijkingen. Als de huid in het donker met Wood's licht (ultraviolet) geïnspecteerd wordt, lichten de vlekken duidelijker op of worden vlekken zichtbaar die eerder niet met het blote oog zijn opgevallen. De poliosis of witte haarlok treedt op als zich een hypomelanotische macula op het hoofd of in de wenkbrauwen bevindt.
Het adenoma sebaceum (AS) wordt bij 50 tot 90 van de volwassen patiënten aangetroffen, mede afhankelijk van de leeftijdsverdeling van de onderzochte groep.5 Deze afwijking wordt als pathognomonisch beschouwd. Klinisch ziet men kleine rood-roze bolhoedvormige, teleangiëctatische papels op de neus en de wangen (figuur 1). De papels kunnen ook bleek zijn, week en (of) vast. De rangschikking is symmetrisch in de nasolabiale plooi, op de wangen, de neus en de kin. Het adenoom kan ontstaan tussen het 3e en 5e jaar of pas in de puberteit.
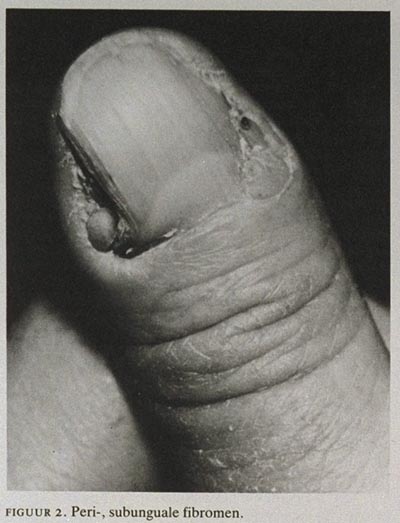
Peri-, subunguale fibromen zijn pathognomonische afwijkingen, die bij ten minste 20 van de volwassen patiënten aangetroffen worden.5 De laesies ontstaan meestal na het 10e levensjaar. De fibromen zijn huidkleurig en bevinden zich onder of naast de nagel en groeien veelal langzaam door (figuur 2). Ze komen vaker voor bij vrouwen dan bij mannen en meer aan de teen-dan aan de vingernagels. Bij slechts één fibroom, zonder andere uitingen van TS, is het verstandig om histopathologisch onderzoek te doen.
De chagrijnplek of ‘peau de chagrin’ is een lederachtig aanvoelende, iets verheven huidkleurige of geel-bruine of oranje plaque die zich meestal lumbosacraal bevindt op de rug.5 De afwijking ontstaat gewoonlijk in de puberteit en komt, soms multipel, bij ten minste 20 van de volwassen patiënten voor.
Huidkleurige, iets bruinrode verdikkingen of fibreuze plaques kunnen gevonden worden op het voorhoofd en het behaarde hoofd. Deze afwijkingen ontstaan waarschijnlijk laat en komen voor samen met het AS. Men vindt ze vaker bij ernstig mentaal geretardeerde patiënten. Toch kunnen ze ook al bij de zuigeling of in de eerste levensjaren voorkomen.
Molluscum fibrosum of ‘skin tag’ is een zacht of gesteeld fibroom, dat in groten getale kan voorkomen in de nek, de oksels, de liezen en ook op de romp en de extremiteiten.5 Ook zijn er kleine papels in de nek en op de romp beschreven.
Tandheelkundige afwijkingen
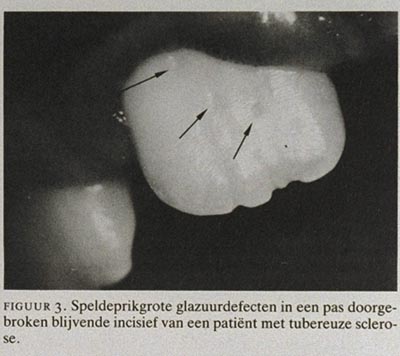
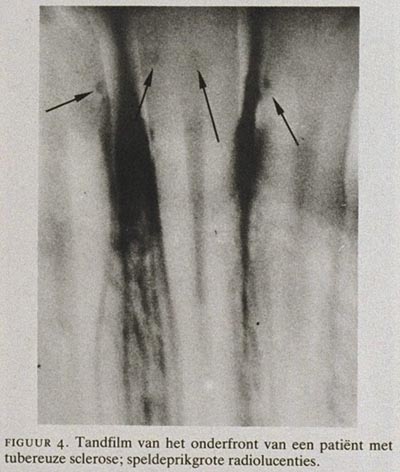
In 1975 is voor het eerst de aandacht gevestigd op het vóórkomen van putjes ter grootte van een speldeprik in het tandglazuur van patiënten met TS.15 Het aantal klinisch waarneembare en met de sonde aftastbare glazuurdefecten op de buccale en linguale vlakken van de gebitselementen varieerde van 1 tot 11. Gemiddeld waren er 3 putjes per vlak te onderscheiden. In de plaats van de glazuurdefecten was geen duidelijk patroon te onderkennen (figuur 3). Op röntgenfoto's (tandfilms) van de frontelementen konden de glazuurdefecten als kleine radiolucenties worden waargenomen (figuur 4). Ook in melkelementen van jonge kinderen met TS zijn deze glazuurdefecten gevonden, zij het wat minder talrijk en relatief kleiner en daardoor moeilijker te diagnostiseren. Het vóórkomen van speldeprikgrote putjes, willekeurig gelokaliseerd in het glazuur van zowel melk- als blijvende elementen bij meerdere patiënten met TS betekent, dat het gaat om een gegeneraliseerd glazuurdefect dat deel uitmaakt van het symptomencomplex TS. De defecten ontstaan gedurende de vorming van de gebitselementen en worden klinisch zichtbaar bij de doorbraak van de elementen.16
Neurologische afwijkingen
Epileptische aanvallen vormen de meest voorkomende reden om een arts te raadplegen. Gegeneraliseerde of partiële insulten komen bij 80-90 van de patiënten voor. TS is de belangrijkste oorzaak van salaamkrampen bij zuigelingen. Ongeveer de helft van de patiënten met TS is mentaal geretardeerd. Er blijkt een duidelijke relatie te bestaan tussen de epilepsie en de retardatie.5 Alle mentaal geretardeerde patiënten hebben ook epilepsie, maar niet alle patiënten met epilepsie zijn mentaal geretardeerd. Hoe eerder de epilepsie voorkomt, hoe groter de kans op (ernstige) mentale retardatie. Dit geldt vooral ook voor het optreden van salaamkrampen bij zuigelingen. Bij een deel van de kinderen met epilepsie in de eerste levensjaren ontwikkelt zich een autistisch gedrag. Kinderen met epilepsie dienen altijd grondig te worden onderzocht op huidafwijkingen passende bij TS. Een kind met salaamkrampen dient volledig onderzocht te worden op de mogelijkheid van TS, tenzij er een andere bekende oorzaak is.
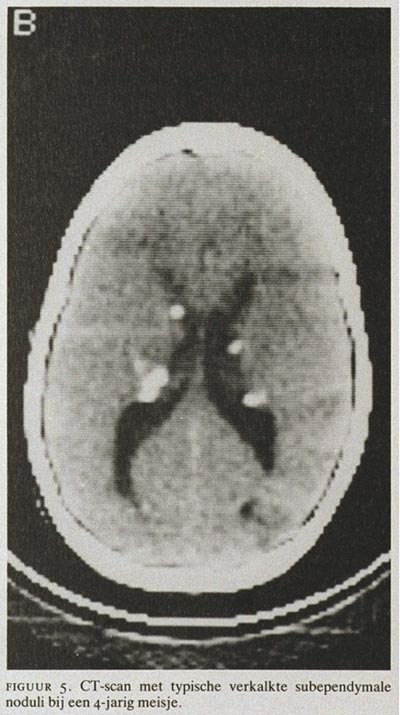
Focale neurologische afwijkingen komen slechts weinig voor. De CT-scan van de schedel toont bij 90 van de TS-patiënten specifieke afwijkingen: subependymale verkalkingen langs de zijventrikels of de 3e ventrikel (figuur 5). Er is geen relatie tussen grootte of aantal van deze verkalkingen en de ernst van de epilepsie of de retardatie. De verkalkingen in de subependymale noduli nemen met de leeftijd toe. Bij enkele procenten van alle patiënten treedt groei op van een nodulus in de omgeving van het foramen van Monro. Dergelijke op de CT-scan aankleurende ‘giant cell astrocytoma's’ kunnen door obstructie van het foramen van Monro een hydrocefalus veroorzaken. In de cortex en de witte stof van grote hersenen en cerebellum kunnen tubera zichtbaar zijn, hypodens (onverkalkt) of hyperdens (verkalkt).17 De CT-scan is superieur in het afbeelden van de subependymale verkalkingen, maar de (sub)corticale hamartomen zijn beter zichtbaar te maken met ‘magnetic resonance imaging’, MRI. Er lijkt wel een relatie te bestaan tussen de (sub)corticale hamartomen aangetoond met CT of MRI en het optreden van epilepsie en retardatie.18
Cardiale afwijkingen
Ongeveer 13 van de patiënten met TS heeft cardiale rabdomyomen. Omgekeerd blijkt 13 van de patiënten met cardiale rabdomyomen TS te hebben. Hoewel het grootste deel van de TS-patiënten met cardiale rabdomyomen hiervan geen klachten ondervindt, treden er bij een klein deel ernstige symptomen op. Echocardiografisch onderzoek is daarom bij iedere TS-patiënt gewenst.
De klinische symptomen zijn in 3 groepen te onderscheiden:
1. Intramurale tumoren kunnen zoveel normaal myocard vervangen door non-contractiel weefsel dat er decompensatie optreedt.
2. Tumoren in het lumen van het hart kunnen de ‘flow’ belemmeren. Meestal leidt dit al kort na de geboorte tot de dood.
3. Intramurale tumoren kunnen ritmestoornissen veroorzaken, zoals atrio-ventriculaire tachycardieën, hartblock, extrasystolen, ventrikelfibrilleren en het Wolff-Parkinson-White-syndroom. Bij therapieresistente ‘epileptische’ aanvallen moet men overwegen of deze geen gevolg zijn van cardiale aritmie.19
Nierafwijkingen
In de nieren kunnen angiomyolipomen (rijk gevasculariseerde tumoren van vet en gladde-spierweefsel) en cysten voorkomen. Hoewel angiomyolipomen veelal asymptomatisch zijn, kunnen de volgende verschijnselen optreden: pijn, hematurie en koorts ten gevolge van een bloeding, soms zelfs met hypovolemische shock; hypertensie, nierinsufficiëntie of een palpabele tumor in de buik. Ultrageluidsonderzoek is een goede screeningsmethode voor TS-patiënten. Angiomyolipomen bij TS-patiënten komen multipel en vrijwel altijd bilateraal voor. Angiomyolipomen bij patiënten zonder TS zijn groter, vrijwel altijd unilateraal en solitair.20 Al dan niet in combinatie met angiomyolipomen kunnen ook multipele cysten in de nieren voorkomen.
Oogheelkundige afwijkingen
De helft van de TS-patiënten heeft retina-hamartomen.21 Deze zijn gewoonlijk asymptomatisch, maar ze vormen wel een belangrijk symptoom om de diagnose TS te kunnen stellen. Er zijn drie typen te onderscheiden:
1. Het meest voorkomende retina-hamartoom is een licht verhoogde afwijking, met een glad oppervlak, zalmkleurig en semi-transparant. Deze afwijkingen zijn vaak over een retinavat gelokaliseerd, hetgeen voor de opsporing van belang is.
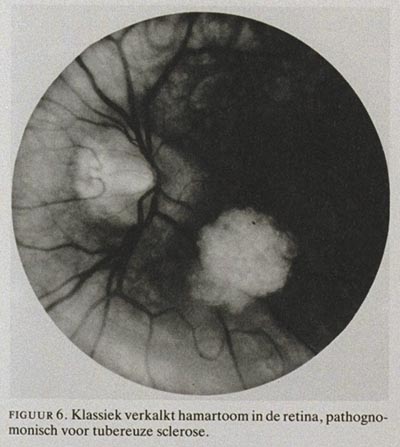
2. Het zgn. klassieke hamartoom is verkalkt en ziet er uit als een moerbei. Deze afwijking is irregulair, wit, verheven en bevindt zich meestal in de buurt van de papil (figuur 6).
3. Het minst frequent is een intermediair type met kenmerken van de eerder genoemde hamartomen.
Minder frequent komen hypomelanotische retinamaculae voor. In lage frequentie is een scala aan andere aspecifieke oogheelkundige afwijkingen beschreven, o.a. colobomen van iris en choroidea, megalocornea en cataract.5
Longafwijkingen
Longafwijkingen zijn zeer zeldzaam (bij 1 van de 100 patiënten met TS) en worden vrijwel alleen gezien bij vrouwelijke volwassen TS-patiënten. De diagnose TS wordt meestal pas overwogen op grond van deze longafwijkingen, daar deze vrouwen meestal weinig of geen andere symptomen hebben. Ze hebben gewoonlijk directe familieleden met klassieke TS. De aandoening wordt gekenmerkt door sterk vergrote longen met vele cysten.
De verschijnselen bestaan uit recidiverende pneumothorax en toenemende dyspnoe. De thoraxfoto toont een toename van het parenchym: honingraat-long. De patiënten overlijden binnen enkele jaren aan een cor pulmonale of pneumothorax.522
Slotbeschouwing
Uit bovenstaande is duidelijk dat vele orgaansystemen kunnen zijn aangedaan, maar dat dit zeer variabel is van patiënt tot patiënt, ook binnen één familie. De diagnose TS is in verband met ‘genetic counselling’ zeer belangrijk. Een eerste kind in een gezin kan slechts geringe verschijnselen hebben, terwijl een volgend kind zeer ernstig is gehandicapt. Een deel van de verschijnselen (zie tabel 1) is pathognomonisch: één hiervan is voldoende voor een zekere diagnose.5 Andere verschijnselen zijn suspect maar niet typisch. Een combinatie van twee of meer van deze verschijnselen (zie tabel 2) maakt de diagnose erg waarschijnlijk maar niet absoluut zeker.5
Gezien de wisselende expressie kan het moeilijk zijn uit te maken of er bij een patiënt al dan niet sprake is van een spontane mutatie. Diagnostische suggesties voor onderzoek van ouders en eventuele andere familieleden worden gegeven in tabel 3. De diagnose TS heeft dus consequenties die zich ver kunnen uitstrekken buiten de patiënt zelf. Hopelijk komen er in de toekomst betrouwbare chromosoommarkers beschikbaar die de nu omslachtige screening vereenvoudigen en het aantal onzekere diagnosen verkleinen.
Literatuur
Wiederholt WC, Gomez MR, Kurtland LT. Incidence andprevalence of tuberous sclerosis in Rochester, Minnesota, 1950 through 1962.Neurology 1985; 35: 600-3.
Recklinghausen F von. Ein Herz von einem Neugeborenenwelches mehrere theils nach aussen, theils na der Höhlen prominerendeTumoren (Myomen) trug. Verh Ges Geburtes, 25 März. MonatsschrGeburtshilfe 1862; 20:1-2.
Bourneville DM. Sclérose tubéreuse descirconvolutions cérébrales: idiotie et épilepsiehémiplégique. Arch Neurol (Paris) 1880; 1: 81-91.
Vogt H. Zur Diagnostik der Tuberösen Sklerose. ZErforsch-Behandl Jugendl Schwachsinns 1908; 2: 1-12.
Gomez MR. Tuberous sclerosis. 2nd ed. New York: RavenPress, 1988.
Fleury P, Groot WP de, Delleman JW, Verbeeten Jr B,Frankenmolen-Witkiezwicz IM. Tuberous sclerosis: the incidence of sporadiccases versus familial cases. Brain Dev 1980; 2: 107-17.
Sampson JR, Scahill SJ, Stephenson JBP, et al. Geneticaspects of tuberous sclerosis in the West of Scotland. J Med Genet 1989; 26:28-31.
Fryer AE, Chalmers A, Connor JM, et al. Evidence that thegene for tuberous sclerosis is on chromosome 9. Lancet 1987; i:659-61.
Connor JM, Pirrit LA, Yates JRW, Fryer AE, Ferguson-SmithMA. Linkage of the tuberous sclerosis locus to a DNA polymorphism detected byv-abl. J Med Genet 1987; 24: 544-6.
Smith M, Haines J, Trofatter J, et al. Linkage studies intuberous sclerosis. Cytogenet Cell Genet 1987; 46: 694.
Northrup H, Beaudet AL, O'Brien WE, Herman GE, LewisRA, Pollack MS. Linkage of tuberous sclerosis to ABO blood group. Lancet1987; ii: 804-5.
Clark RD, Smith M, Pandolfo M, et al. Tuberous sclerosisin a liveborn infant with trisomy due to t (11q23.3; 22q11.2) translocation:is neural cell adhesion molecule a candidate gene for tuberous sclerosis? AmJ Hum Genet 1988; 43: A44.
Hurwitz S, Braverman IM. White spots in tuberoussclerosis. J Pediatr 1970; 77: 578-94.
Oppenheimer EY, Rosman NP, Dooling EC. The lateappearance of hypopigmented macules in tuberous sclerosis. Am J Dis Child1985; 139: 408-9.
Hoff M, Grunsven MF van, Jongbloed WL, 's-GravenmadeEJ. Enamel defects associated with tuberous sclerosis: a clinical andscanning-electron-microscope study. Oral Surg 1975; 40: 261-9.
Hoff M, Grunsven MF van, Poel ACM van de, Anders GJPA.Glazuurdefecten bij tubereuze sclerose: harde feiten van diagnostischebetekenis. Tijdschr Kindergeneeskd 1984; 52: 175-80.
Kimpsley ORE, Kendall BE, Fitz CR. Tuberous sclerosis: aclinico-radiological evaluation of 110 cases with particular reference toatypical presentation. Neuroradiology 1986; 28: 38-46.
Roach ES, Williams DP, Laster DW. Magnetic resonanceimaging in tuberous sclerosis. Arch Neurol 1987; 44: 301-3.
Gibbs JL. The heart and tuberous sclerosis. Anechocardiographic and electrocardiographic study. Br Heart J 1985; 54:596-9.
Baal JG. Angiomyolipoma renis and its relation totuberous sclerosis. Amsterdam, 1987. Proefschrift.
Kiribuchi K, Uchida Y, Fukuyama Y, Maruyama H. Highincidence of fundus hamartomas and clinical significance of a fundus score intuberous sclerosis. Brain Dev 1986; 8: 509-17.
Lieberman BA, Chaimberlain DW, Goldstein RS. Tuberoussclerosis with pulmonary involvement. Can Med Assoc J 1984; 36:287-9.
Artikelinformatie
Citeer dit artikel als




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Reacties