
Samenvatting
- Neuronale migratiestoornissen van de hersenschors vormen een heterogene groep van afwijkingen die worden gekenmerkt door mentale retardatie, epilepsie en hypotonie.
- Ze komen voor bij 1 van de bevolking en bij 20-40 van de onbehandelbare vormen van epilepsie.
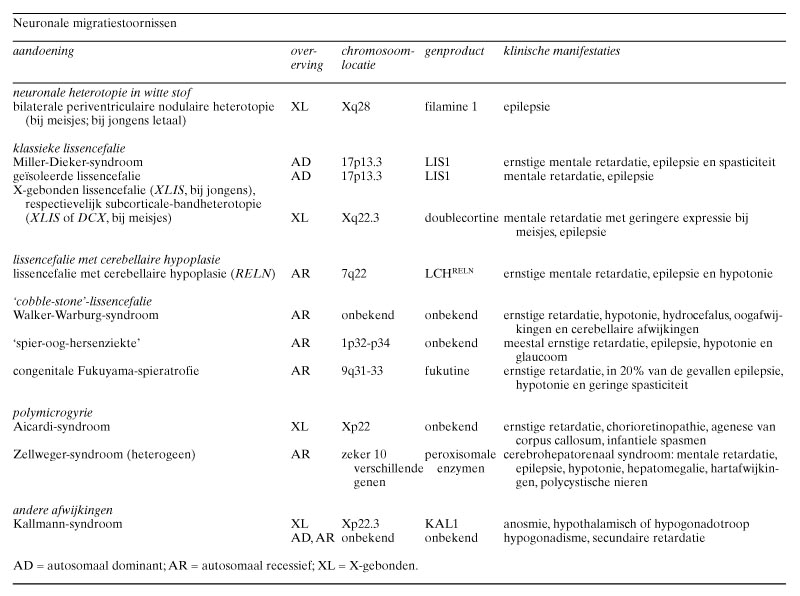
- Stoornissen aan het begin van de migratie leiden tot nodulaire heterotopieën. Bilaterale periventriculaire nodulaire heterotopieën komen familiair voor en zijn X-gebonden. Hierbij zijn corticale neuronen door de afwezigheid van het eiwit filamine 1 niet in staat hun positie aan het ventrikeloppervlak te verlaten.
- De grote groep lissencefalieën kan in een aantal syndromen worden verdeeld, waarbij mutaties in verschillende genen (LIS1, DCX, RELN) leiden tot de voor deze groep kenmerkende agyrie en pachygyrie.
- Een aantal van deze afwijkingen, vooral de kleinere nodulaire heterotopieën en focale corticale dysplasie, komt in aanmerking voor neurochirurgische excisie.
artikel
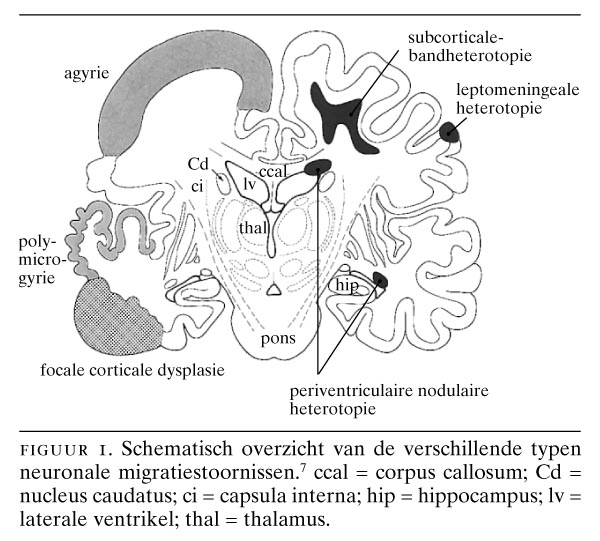
Neuronale migratiestoornissen van de hersenschors zijn ontwikkelingsstoornissen die gepaard kunnen gaan met een trias van klinische manifestaties: mentale retardatie, epilepsie en hypotonie.1 2 Neuronale migratiestoornissen komen bij meer dan 1 van de bevolking voor en bij 20-40 van de onbehandelbare vormen van epilepsie.3 Ze worden gekenmerkt door een aberrante architectuur en een verkeerde differentiatie van de hersenschors.4-8 Hun vóórkomen is wisselend en complex; het gaat om meer dan 25 syndromen. Classificaties zijn gemaakt op grond van ontstaanswijze,1 morfologisch beeld4 9 en vooral beeldvormend neurologisch onderzoek.10 11 Globaal kan de volgende indeling worden gemaakt: (a) agyrie/pachygyrie met de verschillende vormen van lissencefalie (hersenen met een ‘glad’ oppervlak, dat wil zeggen geen of weinig windingen); (b) heterotopieën, zoals de subcorticale-bandheterotopie; (c) polymicrogyrie, en (d) corticale dysplasieën (figuur 1). Veel onderzoek naar de mechanismen van migratiestoornissen werd de laatste jaren verricht aan de ‘reeler’-muis (‘to reel’ = ‘waggelen’), waarbij door de afwezigheid van het eiwit ‘reeline’ vroeg in de ontwikkeling de migratie van neuronen naar de hersenschors stopt.12 13 Inmiddels is hiervan het coderende gen gekloneerd.14 Ook van enkele humane neuronale migratiestoornissen is inmiddels het gen bekend.
In dit derde overzicht over ontwikkeling en ontwikkelingsstoornissen van het humane brein worden de neuronale migratiestoornissen van de grote hersenen behandeld.15 16 Na een kort overzicht van het dierexperimentele werk aan deze stoornissen, bespreken wij aspecten van het symptomenbeeld, beeldvormend onderzoek en neuropathologie van neuronale migratiestoornissen bij de mens. Hierbij is gekozen voor een benadering vanuit het perspectief van de normale neuronale migratie van de hersenschors.15 16
diermodelonderzoeken
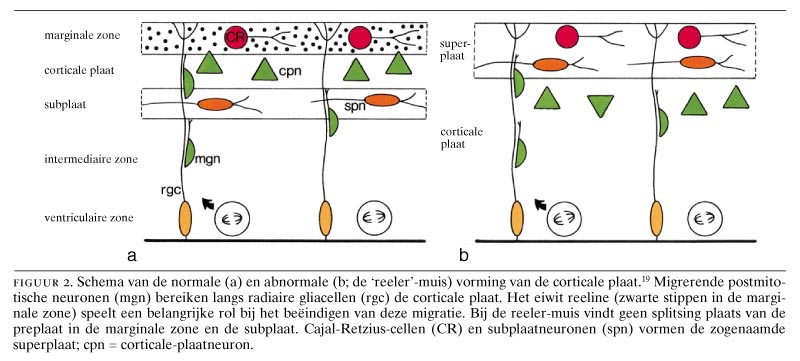
De reeler-muis werd in 1951 min of meer bij toeval ontdekt door Falconer.17 18 Dit is een autosomaal recessieve muizenmutant met afwijkingen in zowel het cerebellum als in de hersenschors. De zogenaamde preplaat, de eerste schorslaag die wordt aangelegd,13 16 19 wordt wel gevormd, maar bij de vorming van de corticale plaat niet gesplitst in de marginale zone en de subplaat (figuur 2). In plaats hiervan blijft de preplaat bestaan als een ‘superplaat’. Dit resulteert in een ‘omgekeerde’ hersenschors, waarbij de vroege populatie neuronen oppervlakkig komt te liggen en de late populatie neuronen in diepere delen van de hersenschors. Ondanks de aberrante lokalisatie van de subplaatneuronen, die een belangrijke rol spelen bij de ingroei van thalamocorticale vezels in de hersenschors, bereiken deze vezels wel de juiste schorsgebieden en de neuronen die overeenkomen met laag IV.20 Ook de corticobulbaire en corticospinale vezelsystemen zijn vergelijkbaar met die in de normale situatie.21 De oorsprongcellen van de piramidebaan zijn echter verspreid door de gehele schors en niet beperkt tot laag V.
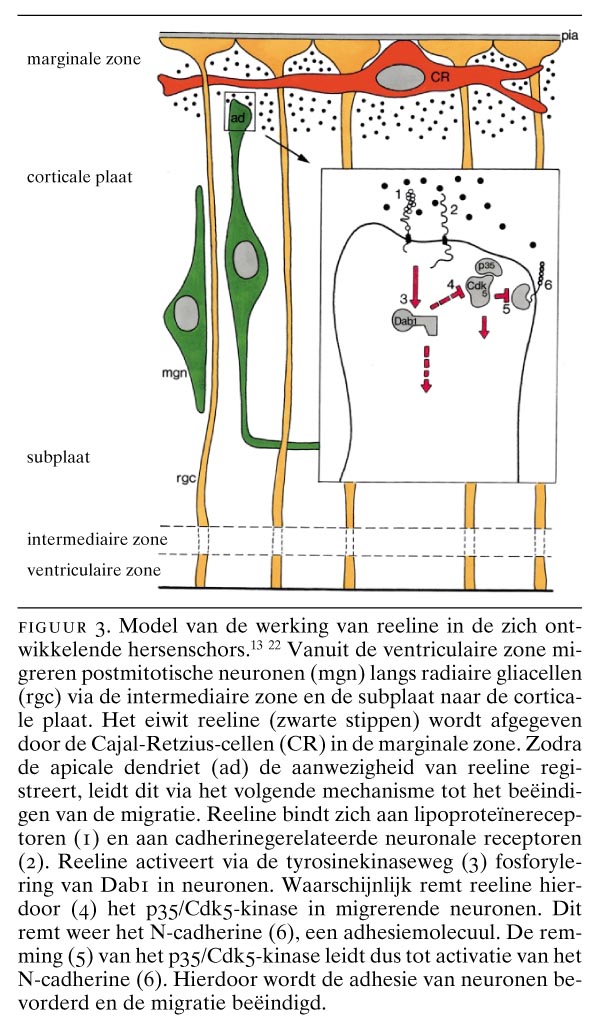
Het eiwit reeline wordt voornamelijk door de Cajal-Retzius-cellen geproduceerd. De mogelijke werking ervan is samengevat in figuur 3. Reeline remt het enzymcomplex p35/Cdk5-kinase in migrerende neuronen. Dit complex remt op zijn beurt de activiteit van het adhesiemolecuul N-cadherine. Remming van het p35/Cdk5-kinase activeert dan ook via het N-cadherine de adhesie tussen neuronen, waardoor de migratie van neuronen wordt gestopt. De reeler-muis vertoont, evenmin als de vergelijkbare ‘scrambler’-mutaties,18 convulsies, een fenomeen dat wel bij de p35-/--mutatie (Cdk5-knock-outmuis) wordt gevonden.7 22 In de hersenschors van zowel p35-/-- als Cdk5-/--muizen kunnen migrerende neuronen de door N-cadherine gemedieerde adhesie niet onderdrukken, waardoor ze niet in staat zijn voorbij de subplaatneuronen te migreren. Het humane reeline-gen (RELN) is gelokaliseerd op chromosoom 7q22 en is nagenoeg identiek aan dat van muizen.23 Onlangs werd er een humane pendant van de reeler-muis gevonden bij een vorm van lissencefalie die gepaard gaat met cerebellaire hypoplasie.24
neuronale migratiestoornissen bij de mens
Voor een aantal neuronale migratiestoornissen begint het duidelijk te worden waar het normale ontwikkelingsproces is verstoord (tabel). Dit geldt voor de lissencefalieën en voor veel heterotopieën. Voor polymicrogyrie en andere corticale afwijkingen is dit echter minder duidelijk. De grote groep lissencefalieën en heterotopieën kan pathogenetisch worden verdeeld in 4 hoofdgroepen:8 (a) stoornissen aan het begin van de migratie (de periventriculaire heterotopieën); (b) stoornissen in het verdere migratieproces (het lissencefalie type 1 en het ‘dubbele-cortex’-syndroom); (c) problemen bij de passage van de subplaat, zoals bij de reeler-muis, en waarschijnlijk ook bij de humane lissencefalievorm die gepaard gaat met cerebellaire hypoplasie; (d) verstoringen van de corticale architectuur, waarbij de hersenschors op meerdere plaatsen wordt onderbroken en een zeer onregelmatig aspect krijgt (‘cobble-stone’- of type-2-lissencefalie). Kenmerkend voor deze heterogene groep van afwijkingen is de trias ernstige mentale retardatie, epilepsie en hypotonie. Een aantal van deze neuronale migratiestoornissen, vooral de kleinere nodulaire heterotopieën en focale corticale dysplasie, komt in aanmerking voor neurochirurgische excisie.25
lissencefalieën en heterotopieën
Periventriculaire heterotopieën
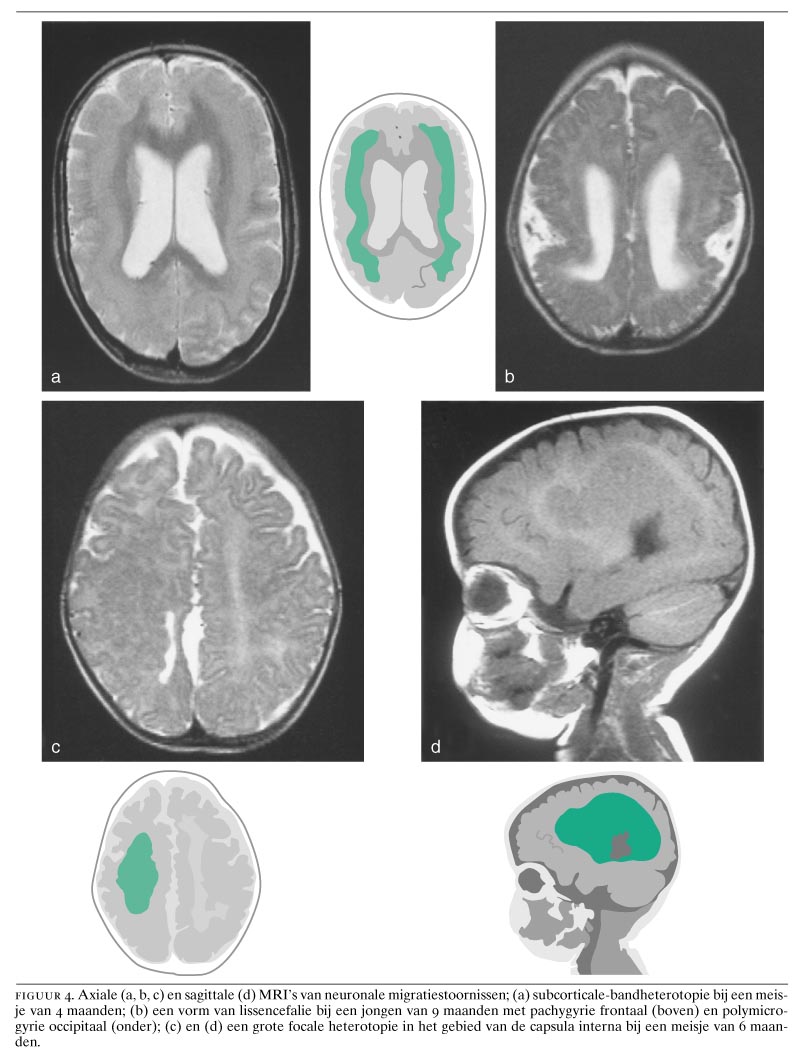
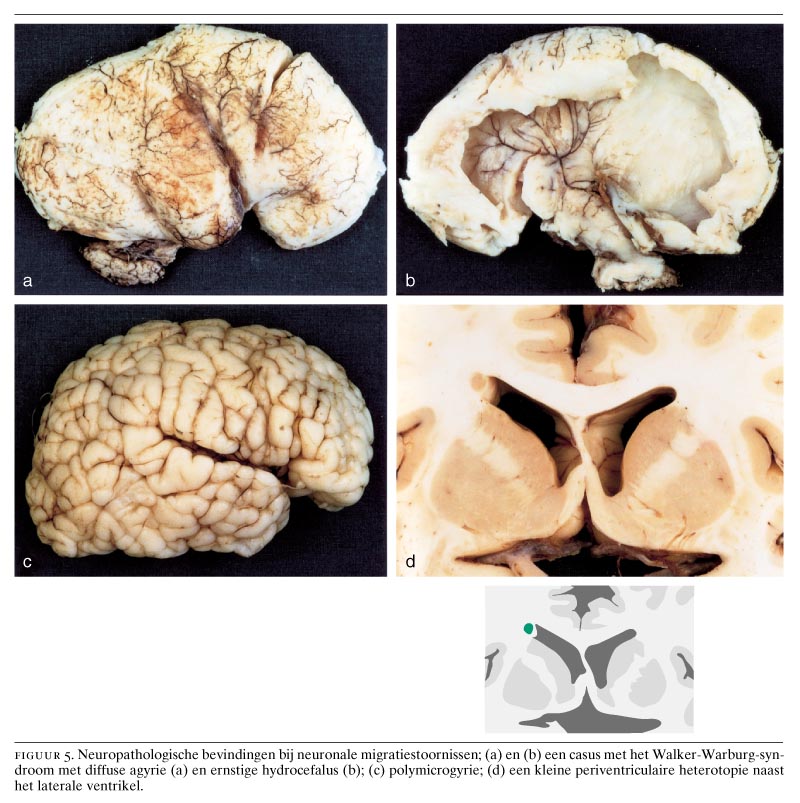
Neuronen van de hersenschors ontstaan in de ventriculaire zone van het telencephalon.16 Hierna treedt migratie op naar de corticale plaat. Bij periventriculaire heterotopieën is een bepaalde populatie neuronen hiertoe niet in staat en worden nodulaire heterotopieën gevormd (zie figuur 1, 4c, 4d en 5d). Deze clusters van neuronen puilen dikwijls uit in de ventrikelholte en zijn met CT- en MRI-onderzoek op te sporen. Bilaterale periventriculaire nodulaire heterotopieën (BPNH) komen familiair voor en zijn X-gebonden.26 Jongens met BPNH-mutaties overlijden meestal al vóór de geboorte.27 Meisjes met BPNH hebben een normale intelligentie. De belangrijkste klinische manifestatie is het optreden van epilepsie, die waarschijnlijk wordt veroorzaakt door de nodulaire heterotopieën. Het syndroom werd gelokaliseerd op Xq28 en het betreffende gen werd geïdentificeerd als filamine 1 (FLN1). FLN1 codeert een actinebindend fosfoproteïne (filamine 1), dat onder andere betrokken is bij de regulatie van de celmigratie.28 Epilepsie bij unilaterale of bilaterale periventriculaire nodulaire heterotopieën kan therapieresistent zijn ten gevolge van de intrinsieke epileptogeniciteit van de periventriculaire heterotopieën.29
Klassieke lissencefalie (type-1-lissencefalie) en het ‘dubbele-cortex-syndroom’
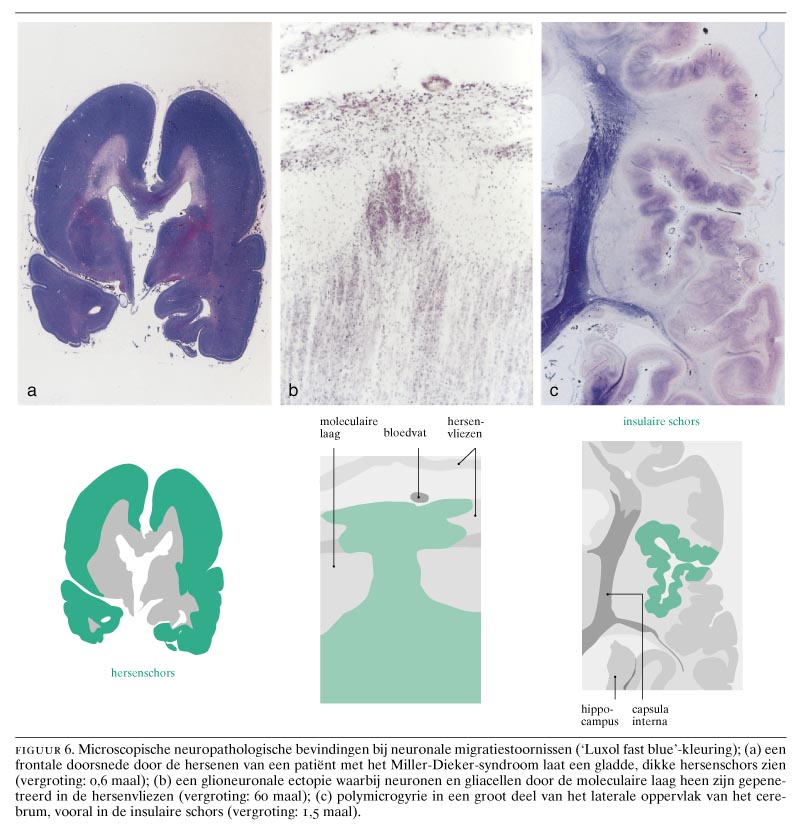
Bij de zogenaamde klassieke lissencefalie hebben de hersenen een overwegend glad oppervlak en een dikke, meestal vierlagige schors. Wanneer karakteristieke craniofaciale afwijkingen, zoals microcefalie, een hoog voorhoofd, een prominent achterhoofd, een brede neusbrug, micrognathie, een lang filtrum en een prominente bovenlip aanwezig zijn, wordt van het Miller-Dieker-syndroom gesproken; zonder deze afwijkingen, vooral aan het aangezicht, spreekt men van een ‘geïsoleerde lissencefalie’.30 De klassieke lissencefalie wordt gekenmerkt door diepe mentale retardatie, epilepsie, voedingsproblemen en een korte levensduur. Bij de klassieke lissencefalie zijn de hersenen meestal kleiner dan normaal. Bij ernstige microcefalie wordt meestal de term ‘microlissencefalie’ gebruikt.9 31 Bij de klassieke lissencefalie vertonen de hersenen een gyrering die varieert van volledige afwezigheid van gyri (‘agyrie’) tot aanwezigheid van een tiental sulci over het gehele brein (‘pachygyrie’). Kenmerkend is de dikke, vierlagige hersenschors (figuur 6a), die van buiten naar binnen de volgende lagen heeft: (a) een moleculaire laag; (b) een dunne oppervlakkige laag van neuronen; (c) een celarme laag, en (d) een opvallend brede laag van neuronen die aan een smalle laag periventriculaire witte stof grenst. Bij het Miller-Dieker-syndroom wordt er meestal een gedeeltelijke deletie van de korte arm van chromosoom 17 (band 17p13.3) gevonden.9 Patiënten met geïsoleerde lissencefalie hebben meestal een minder ernstige graad van lissencefalie dan bij het Miller-Dieker-syndroom.9 10 Deleties of mutaties van tenminste twee genen leiden tot deze klassieke vorm van lissencefalie. Het Miller-Dieker-syndroom en de geïsoleerde vorm van lissencefalie worden veroorzaakt door deleties van het LIS1- of PAFAH1B1-gen.32 Prenatale diagnose van een 17p13.3-deletie is mogelijk aan de hand van een biopsie van chorionvilli met behulp van fluorescentie-in-situhybridisatie en de restrictiefragmentlengte-polymorfismetechniek.
Een tweede, X-gebonden vorm van lissencefalie (XLIS) komt op verschillende wijze bij jongens en meisjes tot expressie.33 34 Bij jongens is het fenotype vergelijkbaar met dat van de klassieke lissencefalie. Wel is er een verschil in lokalisatie: de afwijkingen ten gevolge van LIS1-mutaties komen vooral in de pariëtale en occipitale schorsgebieden tot expressie, terwijl de XLIS-vormen vooral in de frontaalkwab tot uitdrukking komen.34 Meisjes met dit syndroom daarentegen zijn minder ernstig geretardeerd en hebben ook minder ernstige vormen van epilepsie. Zij vertonen een subcorticale-bandheterotopie, waarbij er onder de hersenschors nog een tweede schors aanwezig is (zie figuur 1 en 4a). Er wordt dan ook wel van het ‘dubbele-cortexsyndroom’ gesproken.9 35 Opmerkelijk is dat bij subcorticale-bandheterotopie, ondanks de epileptische activiteit, de subcorticale laminaire heterotopieën wel degelijk ook normale activiteit vertonen, zoals met behulp van functionele MRI werd aangetoond.36 Het gen voor deze X-gebonden vorm van lissencefalie, doublecortine (DCX), dat codeert voor het eiwit doublecortine, is gelokaliseerd op de lange arm van het X-chromosoom op positie Xq22.3.35 Een interessant diermodel voor de subcorticale-bandheterotopie is de tish-mutant (‘telencephalic internal structural heterotopia’) bij de rat.37 Bij deze malformatie bij de rat, die gepaard gaat met convulsies, wordt onder de gewone hersenschors een tweede heterotypische cortex gevormd met vergelijkbare verbindingen als bij de gewone schors.
Lissencefalie met cerebellaire hypoplasie
Bij de reeler-muis zijn, zoals eerder toegelicht, corticale neuronen niet in staat de subplaat te passeren. Bij deze muis worden ook ernstige cerebellaire afwijkingen gevonden. Bij een humaan type lissencefalie, dat gepaard gaat met cerebellaire hypoplasie (LCH), werd onlangs een mutatie van het reeline-gen gevonden op chromosoom 7q22 (LCHRELN).24 Bij MRI-onderzoek bleek de lissencefalie veel minder uitgesproken te zijn dan bij de klassieke vorm van lissencefalie en werd een ernstige hypoplasie van het cerebellum gevonden. Bij de aangedane kinderen is de ontwikkeling van zowel de cognitie als de motoriek ernstig gestoord.
‘Cobble-stone’-lissencefalie (type-2-lissencefalie)
De zogenaamde cobble-stone-lissencefalie of type-2-lissencefalie wordt gekenmerkt door een hobbelig aspect van de hersenschors, een abnormale witte stof, verwijde ventrikels en afwijkingen aan de hersenstam en het cerebellum, waaronder cerebellaire polymicrogyrie.9 De hersenschors is chaotisch gestructureerd door een combinatie van agyrie, pachygyrie en polymicrogyrie met een onregelmatig oppervlak. Van buiten naar binnen kunnen de volgende lagen worden onderscheiden: (a) een brede, buitenste meningeale zone met proliferaties van neuronen en gliacellen in de leptomeningen; (b) een buitenste cellulaire zone met brede onregelmatige ophopingen van neuronen; (c) een diepere cellulaire zone van onregelmatig gerangschikte neuronen; (d) heterotopieën die gedeeltelijk in de witte stof liggen en (e) een binnenste gliazone. Het hersenoppervlak is zeer onregelmatig en lijkt sterk op een straat geplaveid met kasseien. De leptomeningeale heterotopieën worden overigens ook bij andere aangeboren afwijkingen van het centrale zenuwstelsel gevonden, zoals bij het syndroom van Zellweger (zie figuur 6b), bij verkregen destructieve aandoeningen, zoals porencefalie ten gevolge van vasculaire stoornissen of infecties, en bij het foetale alcoholsyndroom.6
De cobble-stone-lissencefalie gaat meestal gepaard met oogmisvormingen en spierdystrofie en komt in verschillende varianten voor.9 Het Walker-Warburg-syndroom wordt gekenmerkt door diffuse agyrie, hydrocefalus, oogafwijkingen en cerebellaire afwijkingen (figuur 5a en 5b). Het is autosomaal recessief en komt over de gehele wereld voor. De precieze lokalisatie van het betreffende gen is nog onbekend. Verwante aandoeningen zijn de ‘spier-oog-hersenziekte’, die alleen in Finland voorkomt en gelokaliseerd werd op chromosoom 1p32-p34,38 en de Fukuyama-spierdystrofie, die vrijwel uitsluitend in Japan voorkomt. Het gen verantwoordelijk voor deze aandoening werd gelokaliseerd op chromosoom 9q31-33 en codeert voor het eiwit fukutine.39 Een interessant muizenmodel voor type-2-lissencefalie wordt gevormd door preseniline-1-deficiënte muizen, waarbij de Cajal-Retzius-cellen verloren gaan.40 Ook hierbij treedt leptomeningeale fibrose op en passeren migrerende neuronen de corticale plaat.
polymicrogyrie
Polymicrogyrie wordt gekenmerkt door de aanwezigheid van talrijke te kleine windingen die gescheiden worden door smalle, ondiepe groeven. De schors is hierbij in geringe mate verdikt met dikwijls neuronale heterotopieën en verwijde ventrikels (zie figuur 5c en 6c). Het macroscopisch beeld suggereert pachygyrie door de accumulatie van talrijke smalle gyri. Meestal is de schors ongelaagd.2 Polymicrogyrie komt voor bij een groot aantal afwijkingen met zeer verschillende oorzaken:1 2 4 6 9 (a) chromosomale afwijkingen; (b) monogenetische afwijkingen, zoals het syndroom van Aicardi; (c) peroxisomale aandoeningen, zoals het syndroom van Zellweger; (d) destructieve laesies, zoals porencefalie ten gevolge van vasculaire stoornissen, vaak in het vascularisatiegebied van de A. cerebri media en na cytomegalovirusinfecties; (e) tweelingschap en (f) overmatig alcoholgebruik tijdens de zwangerschap.
andere corticale afwijkingen
Onder corticale dysplasie wordt verstaan dat op een of meer plaatsen in de hersenschors neuronen en gliacellen op abnormale plaatsen zijn gelegen. Deze afwijking werd beschreven bij dyslexie41 en vooral in de temporaalkwab van patiënten met epilepsie.42 Corticale dysplasie is vaak focaal, maar kan ook op meerdere plaatsen voorkomen,1 43 en kan worden beschouwd als een lichte vorm van een neuronale migratiestoornis. Mogelijk zijn de betreffende neuronen al bij hun ontstaan in de ventriculaire zone afwijkend en hierdoor niet in staat normale lagen te vormen. Men onderscheidt wel focale corticale dysplasie, gegeneraliseerde corticale dysplasie, focale Rolandische macrogyrie, dysplastische afwijkingen van de temporaalkwab en hemimegalencefalie. Een intrigerende migratiestoornis wordt gevonden bij het overigens zeldzame syndroom van Kallmann, dat wordt gekenmerkt door anosmie en infertiliteit. Hierbij bestaat er een stoornis in de migratie van gonadotropine-‘releasing’-hormoon(GnRH)-producerende neuronen vanuit de neusholte naar de hypothalamus. Het eiwit KAL1 ontbreekt bij de X-gebonden vorm van deze aandoening.44 Hoe het KAL1-eiwit de migratie van GnRH-neuronen reguleert, is nog onduidelijk. De genen die betrokken zijn bij de autosomaal dominante en de autosomaal recessieve vormen van deze aandoening zijn nog onbekend. Hormonale substitutietherapie bij patiënten met het Kallmann-syndroom is noodzakelijk om de secundaire geslachtskenmerken te induceren.
conclusies
Parallel aan de snelle vooruitgang in ons begrip van de mechanismen die een rol spelen bij de vorming van de hersenschors, vooral op basis van dierexperimenteel werk, hebben de mogelijkheden van de moderne afbeeldingstechnieken onze kennis van de neuronale migratiestoornissen bij de mens sterk vergroot. Nieuwe neurochirurgische behandelingsmethoden bieden nu soms ruimte voor behandeling van de gevolgen van neuronale migratiestoornissen, vooral de aandoeningen die gepaard gaan met ernstige, niet goed op medicatie reagerende vormen van epilepsie. Correlatie van fenotype en genotype heeft geleid tot nieuwe inzichten in het ontstaan van de hersenschors en de daarbij optredende ontwikkelingsstoornissen. Voor een correcte determinatie van neuronale migratiestoornissen bij een individuele patiënt blijft een zorgvuldige morfologische beschrijving van belang, eens temeer omdat mede op basis van deze determinatie advies gegeven moet worden over prognose, behandeling en herhalingsrisico.
Mw.M.de Leeuw, medisch illustrator, hielp bij het vervaardigen van de illustraties.
Literatuur
Norman MG, McGillivray BC, Kalousek DK, Hill A, PoskittKJ. Congenital malformations of the brain. Pathologic, embryologic, clinical,radiologic and genetic aspects. New York: Oxford University Press;1995.
Harding BN, Copp AJ. Malformations. In: Graham DI, LantosPL, editors. Greenfield's neuropathology. Londen: Arnold; 1996. p.397-533.
Mischel PS, Nguyen LP, Vinters HV. Cerebral corticaldysplasia associated with pediatric epilepsy. Review of neuropathologicfeatures and proposal for a grading system. J Neuropathol Exp Neurol 1995;54:137-53.
Barth PG. Disorders of neuronal migration. Can J NeurolSci 1987; 14:1-16.
Aicardi J. Malformations of the CNS. In: Aicardi J,editor. Diseases of the nervous system in childhood. Clinics in developmentalmedicine. Nr 115/118. Londen: MacKeith Press; 1992. p. 108-202.
Lammens M. Developmental neuropathology in etiology andpathogenesis of human malformations: a personal contribution. Acta BiomedLovaniensia 1997;154:1-175.
Copp AJ, Harding BN. Neuronal migration disorders inhumans and in mouse models - an overview. Epilepsy Res1999;36:133-41.
Gleeson JG, Walsh CA. Neuronal migration disorders: fromgenetic diseases to developmental mechanisms. Trends Neurosci2000;23:352-9.
Dobyns WB, Truwit CL. Lissencephaly and othermalformations of cortical development: 1995 update. Neuropediatrics1995;26:132-47.
Barkovich AJ, Kuzniecky RI, Dobyns WB, Jackson GD, BeckerLE, Evrard P. A classification scheme for malformations of corticaldevelopment. Neuropediatrics 1996;27:59-63.
Kollias SS, Ball WS. In: Ball WS, editor. Pediatricneuroradiology. Philadelphia: Lippincott-Raven; 1997. p. 91-174.
Curran T, D’Arcangelo G. Role of reelin in thecontrol of brain development. Brain Res Rev 1998;26:285-94.
Lambert de Rouvroit C, Goffinet AM. The reeler mouse as amodel of brain development. Adv Anat Embryol Cell Biol1998;150:1-106.
D'Arcangelo G, Nakajima K, Miyata T, Ogawa M,Mikoshiba K, Curran T. Reelin is a secreted glycoprotein recognized by theCR-50 monoclonal antibody. J Neurosci 1997;17:23-31.
Donkelaar HJ ten, Wesseling P, Lammens M, Renier WO,Mullaart RA, Thijssen HOM. Ontwikkeling en ontwikkelingsstoornissen van hethumane brein. I. Vroege ontwikkeling van de grote hersenen.Ned Tijdschr Geneeskd2001;145:345-53.
Donkelaar HJ ten, Wesseling P, Lammens M, Thijssen HOM,Renier WO, Gabreëls FJM. Ontwikkeling en ontwikkelingsstoornissen vanhet humane brein. II. Ontwikkeling van de hersenschors en de grotebaansystemen. Ned Tijdschr Geneeskd2001;145:401-10.
Falconer DS. Two new mutations, trembler and reeler withneurological action in the mouse. J Genet 1951;50:192-201.
Hatten ME. Central nervous system neuronal migration.Annu Rev Neurosci 1999;22:511-39.
Pearlman AL, Faust PL, Hatten ME, Brunstrom JE. Newdirections for neuronal migration. Curr Opin Neurobiol1998;8:46-54.
Molnár Z, Adams R, Goffinet AM, Blakemore C. Therole of the first postmitotic cortical cells in the development ofthalamocortical innervation in the reeler mouse. J Neurosci1998;18:5746-65.
Terashima T. Course and collaterals of corticospinalfibers arising from the sensorimotor cortex of the reeler mouse. Dev Neurosci1995;17:8-19.
Homayouni R, Curran T. Cortical development: Cdk5 getsinto sticky situations. Curr Biol 2000;10:R331-4.
DeSilva U, D’Arcangelo G, Braden VV, Chen J, MiaoGG, Curran T, et al. The human reelin gene: isolation, sequencing, andmapping on chromosome 7 letter. Genome Res1997;7:157-64.
Hong SE, Shugari YY, Huang DT, Al Shahwan S, Grant PE,Hourihane JO, et al. Autosomal recessive lissencephaly with cerebellarhypoplasia is associated with human RELN mutations. Nat Genet2000;26:93-6.
Sisodiya SM. Surgery for malformations of corticaldevelopment causing epilepsy. Brain 2000;123(Pt 6):1075-91.
Huttenlocher PR, Taravath S, Mojtahedi S. Periventricularheterotopia and epilepsy. Neurology 1994;44:51-5.
Eksioglu YZ, Scheffer IE, Cardenas P, Knoll J, DiMario F,Ramsby G, et al. Periventricular heterotopia: an X-linked dominant epilepsylocus causing aberrant cerebral cortical development. Neuron 1996;16:77-87.
Fox JW, Lamperti ED, Eksioglu YZ, Hong SE, Feng Y, GrahamDA, et al. Mutations in filamin 1 prevent migration of cerebral corticalneurons in human periventricular heterotopia. Neuron 1998;21:1315-25.
Sisodiya SM, Free SL, Thom M, Everitt AE, Fish DR,Shorvon SD. Evidence for nodular epileptogenicity and gender differences inperiventricular nodular heterotopia. Neurology 1999;52:336-41.
Dobyns WB, Stratton RF, Greenberg F. Syndromes withlissencephaly. I: Miller-Dieker and Norman-Roberts syndromes and isolatedlissencephaly. Am J Med Genet 1984;18:509-26.
Barkovich AJ, Ferriero DM, Barr RM, Gressens P, DobynsWB, Truwit CL, et al. Microlissencephaly: a heterogeneous malformation ofcortical development. Neuropediatrics 1998;29:113-9.
Reiner O, Carrozzo R, Shen Y, Wehnert M, Faustinella F,Dobyns WB, et al. Isolation of a Miller-Dieker lissencephaly gene containingG protein beta-subunit-like repeats. Nature 1993;364:717-21.
Dobyns WB, Andermann E, Andermann F, Czapansky-Beilman D,Dubeau F, Dulac O, et al. X-linked malformations of neuronal migration.Neurology 1996;47:331-9.
Dobyns WB, Truwit CL, Ross ME, Matsumoto N, Pilz DT,Ledbetter DH, et al. Differences in the gyral pattern distinguish chromosome17-linked and X-linked lissencephaly. Neurology 1999; 53:270-7.
Gleeson JG, Allen KM, Fox JW, Lamperti ED, Berkovic S,Scheffer I, et al. Doublecortin, a brain-specific gene mutated in humanX-linked lissencephaly and double cortex syndrome, encodes a putativesignaling protein. Cell 1998;92:63-72.
Pinard JM, Feydy A, Carlier R, Perez N, Pierot L, BurnodY. Functional MRI in double cortex: functionality of heterotopia. Neurology2000;54:1531-3.
Lee KS, Schottler F, Collins JL, Lanzino G, Couture D,Rao A, et al. A genetic animal model of human neocortical heterotopiaassociated with seizures. J Neurosci 1997;17:6236-42.
Cormand B, Avela K, Pihko H, Santavuori P, Talim B,Topaloglu H, et al. Assignment of the muscle-eye-brain disease gene to1p32-p34 by linkage analysis and homozygosity mapping. Am J Hum Genet1999;64:126-35.
Kobayashi K, Nakahori Y, Miyake M, Matsumura K,Kondo-Iida E, Nomura Y, et al. An ancient retrotransposal insertion causesFukuyama-type congenital muscular dystrophy. Nature1998;394:388-92.
Hartmann D, De Strooper B, Saftig P. Presenilin-1deficiency leads to loss of Cajal-Retzius neurons and cortical dysplasiasimilar to human type 2 lissencephaly. Curr Biol 1999;9:719-27.
Galaburda AM, Sherman GF, Rosen GD, Aboitiz F, GeschwindN. Developmental dyslexia: four consecutive patients with cortical anomalies.Ann Neurol 1985;18:222-33.
Hardiman O, Burke T, Phillips J, Murphy S, O'MooreB, Staunton H, et al. Microdysgenesis in resected temporal neocortex:incidence and clinical significance in focal epilepsy. Neurology1988;38:1041-7.
Meencke HJ. Diagnosis of cortical and subcorticaldysplasias in epilepsy. In: Schmidt D, Schachter SC, editors.Epilepsy-problem solving in clinical practice. Londen: Dunitz; 2000. p.95-109.
Franco B, Guioli S, Pragliola A, Incerti B, Bardoni B,Tonlorenzi R, et al. A gene deleted in Kallmann's syndrome shareshomology with neural cell adhesion and axonal path-finding molecules. Nature1991;353:529-36.
Artikelinformatie
Citeer dit artikel als




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Reacties