
artikel
Inleiding
de ziekte
Het Wolfram-syndroom is een progressieve neurodegeneratieve afwijking, die voor het eerst werd beschreven in 1938 door Wolfram en Wagener.1 Het is een autosomaal recessief overervende aandoening, waarbij in ongeveer 25 van de betreffende families consanguïniteit voorkomt.2 De diagnostische minimumcriteria zijn juveniele insulineafhankelijke diabetes mellitus en opticusatrofie. Deze kenmerken manifesteren zich meestal in het eerste decennium. In het tweede decennium wordt bij veel van de aangedane personen ook centrale (hypothalame) diabetes insipidus (73) en slechthorendheid van de hoge tonen (62) gevonden.3 Een gangbaar acroniem is ‘DIDMOAD-syndroom’ (‘diabetes insipidus, diabetes mellitus, opticusatrofie en doofheid’). Additionele ziektekenmerken die in latere decennia kunnen optreden zijn: dilatatie van de urinewegen (58), neurologische complicaties (cerebellaire ataxie en/of myoclonus in 62 van de gevallen), gastro-intestinale motiliteitsstoornissen (24) en primaire gonadale atrofie (70).3 Uiteenlopende psychiatrische stoornissen komen bij ongeveer 60 van de patiënten op volwassen leeftijd voor; 25 van deze patiënten wordt daarvoor tijdelijk of permanent opgenomen in een psychiatrische instelling.4 Patiënten met het Wolfram-syndroom hebben een beperkte levensverwachting, met een mediane levensduur van ongeveer 30 jaar (uitersten: 25-49).5 Patiënten overlijden veelal aan een centrale respiratoire insufficiëntie ten gevolge van hersenstamatrofie.5 De heterozygote dragers van een genmutatie in het WFS1-gen hebben een statistisch significant hoger risico op gehoorverlies, diabetes mellitus en psychiatrische stoornissen dan niet-dragers.6 7
het gen
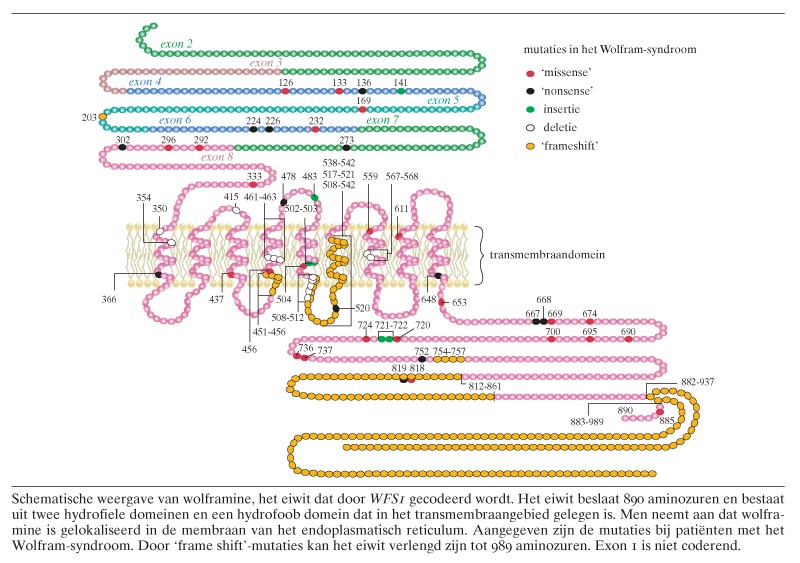
Het is aangetoond dat mutaties in het WFS1-gen kunnen leiden tot het Wolfram-syndroom.8 9 Het WFS1-gen is gelegen op chromosoom 4p16 en beslaat 33,4 kb. Het bestaat uit 8 exonen, waarvan exon 1 niet-coderend is. Tot op heden zijn er tientallen mutaties (deleties, inserties, ‘nonsense’- en ‘missense’-mutaties) beschreven die het syndroom kunnen veroorzaken. In exon 8, het grootste exon, worden de meeste pathogene mutaties gevonden (figuur).10 Het merendeel is inactiverend en er is geen duidelijke relatie tussen de aard en positie van de mutaties (genotype) en de ernst van de genoemde kenmerken (fenotype).
Behalve tot het Wolfram-syndroom blijken heterozygote missense-mutaties in WFS1 ook te leiden tot een niet-syndromale, autosomaal dominant overervende perceptieve slechthorendheid type 6 of 14.11 Daarnaast is er een tweede locus (WFS2) voor het Wolfram-syndroom beschreven voor 3 Jordaanse families, dat is gelegen op chromosoom 4q22-24. Opvallend aan deze vorm van het Wolfram-syndroom is dat geen van de aangedane personen diabetes insipidus krijgt.12
het eiwit
WFS1 codeert voor het polypeptide wolframine, dat bestaat uit 890 aminozuren met een molecuulgewicht van 100 kDa. Het is een transmembraaneiwit met drie typische structurele domeinen: een hydrofiel N-terminaal domein van ongeveer 300 en een hydrofiel C-terminaal domein van ongeveer 240 aminozuren, met daartussen een 350 aminozuren groot hydrofoob fragment waarin de 9 transmembraan-?-helices gelokaliseerd zijn (zie de figuur). De subcellulaire lokalisatie van wolframine in het endoplasmatisch reticulum (ER) zou erop kunnen duiden dat het een rol speelt in transmembraanprocessen, de productie van eiwitten en/of de regulatie van de calciumhomeostase in het ER.13 Wolframine vertoont geen enkele gelijkenis met bestaande eiwitten in publieke databases en lijkt als zodanig lid te zijn van een nieuwe eiwitfamilie.
de cel
Tot op heden zijn de exacte rol en functie van wolframine in het menselijk lichaam nog onduidelijk. Wolframine speelt waarschijnlijk een cruciale rol in de overleving van zowel bètacellen in de pancreas als van bepaalde neuronen in het centraal zenuwstelsel.13 Het werkingsmechanisme is echter nog niet bekend. Wolframine komt sterk tot expressie in het hart, gemiddeld tot expressie in de hersenen, placenta, longen en pancreas, en zwak tot expressie in lever, skeletspieren en nieren.9
Wolframine wordt met name gevonden in speciale neuronen in de hippocampus, in de nabijheid van het corpus amygdaloideum, en de oppervlakkige laag van de allocortex. Deze expressiegebieden zijn met name gerelateerd aan het limbische systeem en kunnen bijdragen aan de psychiatrische stoornissen die bekend zijn bij het Wolfram-syndroom.13
de populatie
Het Wolfram-syndroom is zeldzaam. In een nationale studie in Groot-Brittannië bleken er 45 patiënten met het Wolfram-syndroom te zijn. De prevalentie in Groot-Brittannië is derhalve 1:770.000 en het dragerschapsrisico is 1:354.3 Momenteel zijn er, voorzover ons bekend, 9 families met 13 in leven zijnde patiënten met het Wolfram-syndroom in Nederland. Voor het merendeel van deze families zijn reeds mutaties in WFS1 gevonden. Daarnaast hebben wij in de afgelopen 25 jaar nog 4 andere families met 7 Wolfram-syndroompatiënten gekend, echter, deze zijn inmiddels allen overleden. Het geringe aantal patiënten wordt mogelijk ook veroorzaakt door de relatieve onbekendheid van dit zeldzame beeld onder artsen.
diagnostiek
Voor de diagnose van het Wolfram-syndroom dient minimaal juveniele insulineafhankelijke diabetes mellitus en opticusatrofie vóór het 15e levensjaar vastgesteld te worden. Op basis van deze criteria worden bij 90 van de patiënten met het Wolfram-syndroom mutaties in WFS1 gevonden in tenminste één van beide allelen.10 Additionele kenmerken, zoals diabetes insipidus, slechthorendheid, renale, gastro-intestinale, neurologische en psychiatrische stoornissen, leveren geen verbetering in diagnostische precisie op.10 DNA-analyse van WFS1 dient vooralsnog louter ter bevestiging van de op klinische gronden gestelde diagnose ‘syndroom van Wolfram’. Prenatale diagnostiek is hiermee ook mogelijk. De afwezigheid van enkele veelvoorkomende mutaties dan wel ‘hot spots’ of clusteringen, alsmede de aanwezigheid van vele unieke mutaties maken het noodzakelijk het gehele coderende gebied van WFS1 te analyseren. DNA-diagnostiek van het WFS1-gen kan vanaf medio 2002 verricht worden op de afdeling Anthropogenetica van het Universitair Medisch Centrum St Radboud te Nijmegen. Aan het klinisch-chemisch laboratorium van de Isala Klinieken, locatie Weezenlanden, te Zwolle en aan de afdeling Medische Genetica te Antwerpen wordt wetenschappelijk onderzoek verricht naar het WFS1-gen.
Literatuur
Wolfram D, Wagener H. Diabetes mellitus and simple opticatrophy among siblings: report of four cases. Mayo Clin Proc1938;13:715-8.
Cremers CWRJ, Wijdeveld PGAB, Pinckers AJLG. Juvenilediabetes mellitus, optic atrophy, hearing loss, diabetes insipidus, atonia ofthe urinary tract and bladder, and other abnormalities (Wolfram syndrome). Areview of 88 cases from the literature with personal observations on 3 newpatients. Acta Paediatr Scand 1977;264 Suppl:1-16.
Barrett TG, Bundey SE, Macleod AF. Neurodegeneration anddiabetes: UK nationwide study of Wolfram (DIDMOAD) syndrome. Lancet1995;346:1458-63.
Swift M, Swift RG. Psychiatric disorders and mutations atthe Wolfram syndrome locus. Biol Psychiatry 2000;47:787-93.
Fuqua JS. Wolfram syndrome: clinical and genetic aspects.Endocrinologist 2000;10:51-9.
Ohata T, Koizumi A, Kayo T, Shoji Y, Watanabe A, Monoh K,et al. Evidence of an increased risk of hearing loss in heterozygous carriersin a Wolfram syndrome family. Hum Genet 1998;103:470-4.
Swift RG, Perkins DO, Chase CL, Sadler DB, Swift M.Psychiatric disorders in 36 families with Wolfram syndrome. Am J Psychiatry1991;148:775-9.
Inoue H, Tanizawa Y, Wasson J, Behn P, Kalidas K,Bernal-Mizrachi E, et al. A gene encoding a transmembrane protein is mutatedin patients with diabetes mellitus and optic atrophy (Wolfram syndrome). NatGenet 1998;20:143-8.
Strom TM, Hörtnagel K, Hofmann S, Gekeler F, ScharfeC, Rabl W, et al. Diabetes insipidus, diabetes mellitus, optic atrophy anddeafness (DIDMOAD) caused by mutations in a novel gene (wolframin) coding fora predicted transmembrane protein. Hum Mol Genet 1998;7:2021-8.
Khanim F, Kirk J, Latif F, Barrett TG. WFS1/wolframinmutations, Wolfram syndrome, and associated diseases. Hum Mutat (Online)2001;17:357-67.
Bespalova IN, Van Camp G, Bom SJH, Brown DJ, Cryns K,DeWan AT, et al. Mutations in the Wolfram syndrome 1 gene (WFS1) are a commoncause of low-frequency sensorineural hearing loss. Hum Mol Genet2001;10:2501-8.
El-Shanti H, Lidral AC, Jarrah N, Druhan L, Ajlouni K.Homozygosity mapping identifies an additional locus for Wolfram syndrome onchromosome 4q. Am J Hum Genet 2000;66:1229-36.
Takeda K, Inoue H, Tanizawa Y, Matsuzaki Y, Oba J,Watanabe Y, et al. WFS1 (Wolfram syndrome 1) gene product: predominantsubcellular localization to endoplasmic reticulum in cultured cells andneuronal expression in rat brain. Hum Mol Genet2001;10:477-84.
Artikelinformatie



{kind=link}
Reacties