
Inleiding
de ziekte
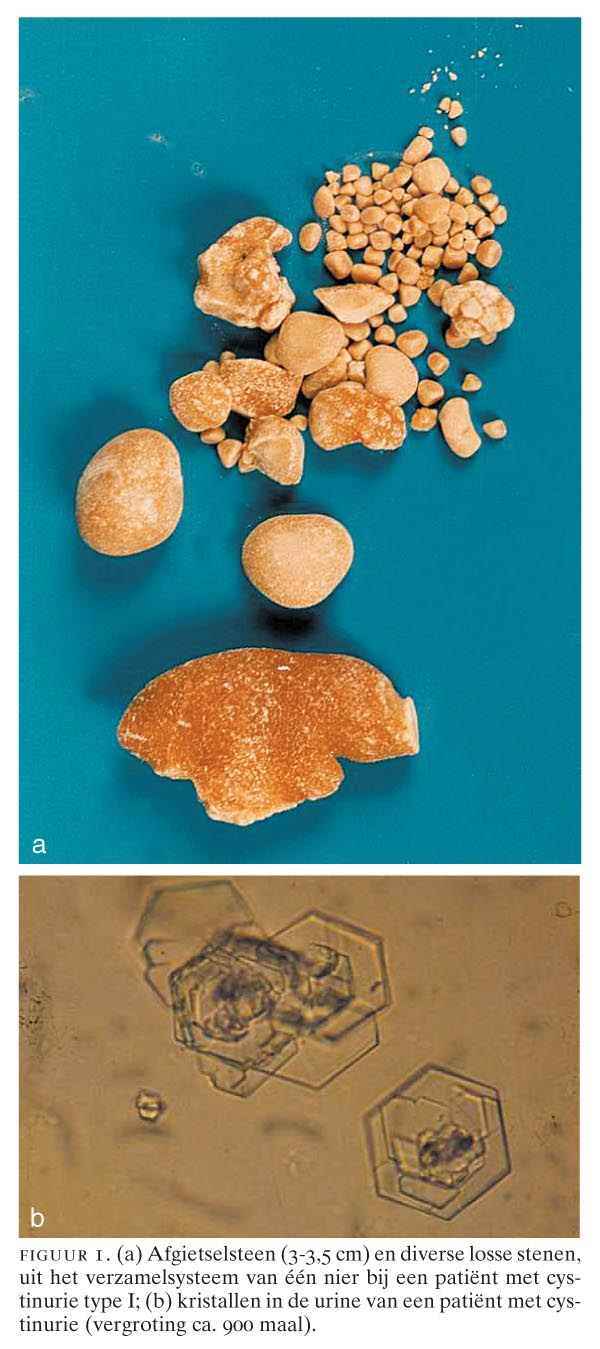
Nierstenen komen veel voor en kunnen worden veroorzaakt door hyperexcretie van slecht oplosbare moleculen, zoals calcium, oxalaat, cystine en uraat. Cystinestenen zijn goudgeel tot bruin van kleur en kunnen in grootte variëren van zandkorrels tot afgietsels van het nierbekken (figuur 1a). Cystinurie is een stoornis in de terugresorptie van cystine in de nier. Ongeveer eenderde van de symptomatische patiënten met cystinurie heeft de eerste steen al vóór het 10e levensjaar, eenderde tussen 10 en 20 jaar en eenderde daarna. De patiënt presenteert zich meestal met een niersteenkoliek en soms met een secundair ontstane urineweginfectie. Mannen hebben vaker en ernstiger klachten dan vrouwen; de symptomen zijn zeer variabel. Sommige patiënten met cystinurie hebben uitgebreide steenvorming (zie figuur 1a), terwijl een broer of zuster met een even hoge cystine-uitscheiding in de urine geen of nauwelijks klachten heeft. Het is niet bekend waardoor deze verschillen worden veroorzaakt…
Artikelinformatie
Citeer dit artikel als



{kind=link}
(Geen onderwerp)
Nijmegen, maart 2003,
Zoals Breuning en Hamdy aangeven (2003:245-7), is het verband genotype-fenotype niet eenduidig. Recent werd zelfs aangetoond dat mutaties in het ‘solute carrier 7, member 9’(SCL7A9)-gen kunnen voorkomen bij de 3 typen van cystinurie.1 Met het behandelingsadvies van de schrijvers hebben wij problemen. De hoeksteen van de behandeling is naar hun mening de vergroting van de vochtinname tot dagelijks 3,5-4 l per 24 uur om de pH van de urine hoog te houden. Dit is onzes inziens onjuist. Voldoende vochtopname is noodzakelijk om de cystineconcentratie in de urine beneden de oplosbaarheidsgrens te houden. In het verleden is veel onderzoek verricht naar de invloed van waterdiurese op de urine-pH. Uitvoerig gebeurde dit door onder anderen Reid en Hills.2 3 Wanneer er werd uitgegaan van een bicarbonaatrijke urine (pH > 6,6), treedt er een daling van de urine-pH op. Wanneer vóór de waterdiurese de urine-pH laag is (pH < 6,6), stijgt de urine-pH. In hun artikel geven de schrijvers hiervoor een verklaring.
De aanbevolen vochthoeveelheid is niet steeds voldoende. Bij homozygote mannen bedraagt de cystine-excretie 551-2200 mg (n = 18) en bij homozygote vrouwen 675-1922 mg (n = 17).4 Bij een urine-pH van 7 is, zoals duidelijk is uit de figuur, de aanbevolen vochthoeveelheid onvoldoende. Bij het advies dient men zich te realiseren dat de vochtopname niet gelijk is aan de diurese, gezien de perspiratio insensibilis. Tevens dient men rekening te houden met het dag- en nachtritme voor water en elektrolyten. Bij eenzelfde inname van vocht en elektrolyten is de urine gedurende de nacht meer geconcentreerd en zuurder. Het is dus vooral gedurende de nacht dat een neerslag van cystine kan ontstaan.
Voor het instellen van de behandeling nemen wij de patiënten in de kliniek op en stellen de benodigde vochthoeveelheid verdeeld over 24 uur vast. Tevens zorgen wij voor een pH-verhoging van de urine.5 Volmaakt is dit niet, mede omdat de pH in de urine in de blaas hoger is dan in de verzamelbuis. Ten gevolge van het ontbreken van carboanhydrase in het lumen van de verzamelbuis stijgt de concentratie van H2CO3. Bij het ontbreken van carboanhydrase is de omzetting van H2CO3 in CO2 en H2O vertraagd. Dit leidt tot een pH-verhoging, onder andere in de blaas.6 Voor een levenslange behandeling dienen adequate richtlijnen te worden gegeven. Wanneer wij twijfelen aan de medewerking van de patiënten, trachten wij in ieder geval 's nachts een voldoende diurese te bereiken.
De auteurs vermelden tevens dat stoffen worden toegediend die een verbinding aangaan met cystine: penicillamine of tiopronine. Deze stoffen hebben zeker een plaats bij een verder gevorderd stadium van cystinurie met multipele steenvorming. Deze stoffen kunnen evenwel ernstige bijwerkingen hebben. Na penicillaminetoediening krijgt bijna 25% van de patiënten een epimembraneuze glomerulonefritis. Daarnaast kunnen leukopenie, trombocytopenie, ‘rash’ en verlies van smaak optreden. De bijwerkingen van tiopronine zouden volgens de meeste onderzoekers minder frequent optreden.
Leclerc D, Boutros M, Suh D, Wu Q, Palacin M, et al. SLC7A9 mutations in all three cystinuria subtypes. Kidney Int 2002;62:1550-9.
Reid EL, Hills AG. Diffusion of carbon dioxide out of the distal nephron in man during antidiuresis. Clin Sci 1965;28:15-28.
Woeber KA, Reid EL, Kiem I, Hills AG. Diffusion of gases out of the distal nephron segment in man. J Clin Invest 1963;42:1689-704.
Evans WP, Resnick MI, Boyce WH. Homozygous cystinuria – evaluation of 35 patients. J Urol 1982;127:707-9.
Monnens LA, Noordam K, Trijbels F. Necessary practical treatment of cystinuria at night. Pediatr Nephrol 2000;14:1148-9.
DuBose jr TD. Hydrogen ion secretion by the collecting duct as a determinant of the urine to blood PCO2 gradient in alkaline urine. J Clin Invest 1982;69:145-56.
(Geen onderwerp)
Leiden, april 2003,
Wij zijn het eens met het commentaar van collega's Geelen et al. betreffende de relatie tussen vochtinname en pH-veranderingen, maar niet met hun suggestie om het gebruik van sulfhydrylderivaten te beperken tot uitsluitend patiënten met multipele steenvorming. Naar onze mening komt de behandeling dan te laat in het natuurlijke beloop van de ziekte.
Wij hadden niet de intentie in ons artikel een uitgebreid behandelingsadvies te geven. De beknopte wijze waarop het beleid in het artikel geformuleerd is, kan echter tot verwarring leiden. Daarom willen wij in het navolgende onze mening over de therapie bij cystinuriepatiënten uitgebreider weergeven.
Door de lage oplosbaarheid van cystine hebben mensen met cystinurie voortdurend een hoog risico op recidiverende nierstenen. Het risico op nierfunctiestoornissen is eveneens hoog. Ruim 70% van de patiënten met cystinurie heeft tekenen van verminderde nierfunctie, hoewel bij slechts 5% van hen dialyse nodig is. Preventieve behandelingen van steenvorming zijn effectief gebleken ter bescherming van de nierfunctie.1 2
Cystine is slecht oplosbaar en de oplosbaarheid is pH-afhankelijk. Een aanzienlijke vochtinname is dus wenselijk dag en nacht, met als doel een urine-output van tenminste 3 l per dag te bereiken. Om de oplosbaarheid verder te optimaliseren is een urine-pH tussen de 7,5 en 8 noodzakelijk. Dit wordt bij het merendeel van de patiënten bereikt met natriumbicarbonaat en/of kaliumcitraat. Daardoor wordt de oplosbaarheid van cystine in de urine verhoogd en cystinekristalvorming voorkomen.
Bij meer dan tweederde van de patiënten, met name homozygoten, schieten deze maatregelen echter tekort en blijft de uitscheiding van cystine in de urine boven de 250 mg/dag. Het gevaar van steenvorming blijft dan hoog. In deze gevallen wordt tweedelijnstherapie met het sulfhydrylderivaat tiopronine aanbevolen.3 4 Dit middel heeft een sulfhydrylgroep, die de S-S-binding in cystine kan openbreken, hetgeen leidt tot een beter oplosbaar complex met het cysteïnemonomeer. Tiopronine blijkt goed te worden verdragen, met geringe bijwerkingen bij 30% van de patiënten, en effectief te zijn in het verminderen van de frequentie van steenvorming, met name in de moeilijk te behandelen gevallen, waar aanzienlijke hydratie en het alkaliniseren van de urine gefaald hebben.5 Het oudere, vergelijkbaar effectieve preparaat, penicillamine, is van de markt gehaald wegens de vele ernstige bijwerkingen.4
Captopril, een eerstegeneratie-angiotensine-‘converting’-enzymremmer, vermindert via hetzelfde mechanisme als tiopronine ook de cystine-excretie en is de eerste keuze voor de behandeling van hypertensie bij cystinuriepatiënten.6 Ook wordt een methionine- en natriumbeperkt dieet aanbevolen om cystine-uitscheiding in de urine te verminderen.7
In de praktijk blijkt echter dat de meeste cystinuriepatiënten het strenge regime van aanzienlijke hydratie en optimale alkalinisatie van de urine niet kunnen volhouden, zodat een aanvullende behandeling met tiopronine noodzakelijk is, teneinde steenvorming te beperken en daarmee nierfunctieverlies te voorkomen.
Lindell A, Denneberg T, Granerus G. Studies on renal function in patients with cystinuria. Nephron 1997;77:76-85.
Gambaro G, Favaro S, D'Angelo A. Risk for renal failure in nephrolithiasis. Am J Kidney Dis 2001;37:233-43.
Dahlberg PJ, Berg CJ van den, Kurtz SB, et al. Clinical features and management of cystinuria. Mayo Clin Proc 1977;52:533-42.
Linari F, Marangela M, Fruttero B, Bruno M. The natural history of cystinuria: a 15 year follow-up in 106 patients. In: Smith LH, Robertson WG, Finlayson B, editors. Urolithiasis, clinical and basic research. New York: Plenum Press; 1981. p. 145-9.
Pak CYC, Fuller C, Sakhaee K, Zerwekh JE, Adams BV. Management of cystine nephrolithiasis with alpha-mercaptopropionylglycine. J Urol 1986;136:1003-8.
Cohen TD, Streem SB, Hall P. Clinical effect of captopril on the formation and growth of cystine calculi. J Urol 1995;154:164-6.
Ng CS, Streem SB. Contemporary management of cystinuria. J Endourol 1999;13:647-51.